Huntington's chorea (Huntington's) is a chronic autosomal dominant neurodegenerative disease characterized by onset in middle age. It is a progressive impairment of cognitive functions, combined with the appearance of involuntary movements and impaired coordination. As a result, this disease combines symptoms of choreic hyperkinesis and dementia.
The pathological condition was first described in 1872 by George Huntington. The incidence of Huntington's chorea is 9 cases per 100 thousand people. The first symptoms of the disease are observed by people aged 35-40 years. However, cases of the disease occurring at 3 years of age and even at 80-90 years of age have been reported. The disease is characterized by uneven geographical and ethnic distribution. It is rare in Asian populations. The pathology is more common in some isolated regions, for example, the island of Tasmania.
Description
Huntington's chorea - what kind of disease, what is its essence? It is a serious hereditary disease with typical mental and physical symptoms that usually occur between 30 and 45 years of age. But signs may appear earlier. If they are observed around the age of 20, the pathology is referred to as juvenile Huntington's disease (Westphal variant). As a rule, it is characterized by uncontrollable twitching of the limbs, torso, and dizziness. The disease leads to psychological personality disorder and dementia.
Huntington's syndrome affects about 5-10 people per 100,000. Unfortunately, it cannot be prevented, slowed, or cured. Life expectancy is reduced not by the disease itself, but by a weakened immune system.
The pathology got its name in honor of the American physician George Huntington, who first described it in 1872.
Causes
Genetic aspects . Increase in the number of CAG repeats - triplets of the huntingtin gene (143100, HD gene, IT15, 4p16-3, Â). Paternal transmission of the gene, as well as an increase in the number of repeats, leads to a more severe form of the disease, early onset and rapid progression.
Pathogenesis . Progressive death of nerve cells. A marked decrease in the content of neurotransmitters (g - aminobutyric acid, glutamate decarboxylase, substance P, enkephalins) in the basal ganglia. A marked decrease in the activity of the mitochondrial respiratory chain in the caudate nucleus.
Pathomorphology . Macroscopically: atrophy of the caudate nucleus and dilatation of the ventricles. Microscopically: gliosis and neuronal death, especially in the caudate nucleus and putamen.
Causes and risk factors
The cause of the disease is a change (mutation) in the IT15 gene on the 4th chromosome.
The basis of the mutation in Huntington's disease is the proliferation of the CAG (cytosine-adenine-guanine) triplet. The normal state is up to 35 CAG triplets; with a number of 36-39, the prognosis is unclear; with more than 40 triplets, a person will most likely become ill with this insidious disease. The rule “the more CAG triplets, the faster the disease outbreak” is especially true for people with an extreme number of CAG triplets.
A gene containing fewer than 36 triplets produces the protein huntingtin, which is essential for proper brain development. Huntington's disease is a disease with an autosomal dominant pattern of inheritance. This means that it occurs mostly in every generation. If one parent has this disorder, there is a 50% risk that the child will also develop the disorder.
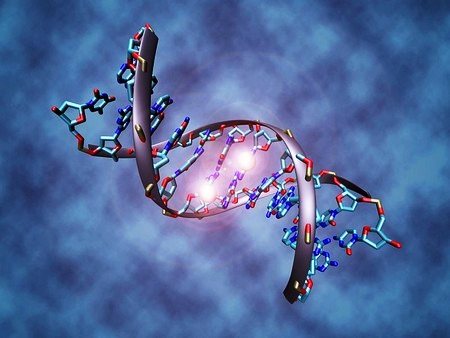
When diagnosing Huntington's disease, it is necessary to take into account that the presence of a mutation carrier in the family is not always known. Reasons: premature death from another pathology, divorce, unknown father. New mutations are very rare.
Patients suffering from the disease are advised not to have children. But the problem is that in most cases it is diagnosed only after the baby is born. The genetic mutation is present from birth but usually begins to appear in adulthood.
How long do people live with Huntington's disease?
Life expectancy with Huntington's disease is 15-20 years.
Patients in most cases die from associated diseases, cachexia, and there have also been cases of suicide. In the juvenile form, degenerative changes develop faster; patients live no more than 10 years.
If the pathology begins in old age, which happens very rarely, the rate of development of the disease is very slow, and dementia practically does not occur. This course of chorea is observed with maternal transmission of the mutant gene.
· Apr 22, 2019
Symptoms
A person usually comes to the doctor because of problems with movement, accompanied by mental changes. Symptoms and signs of Huntington's disease most often appear around age 40. The clinical picture gradually worsens as a result of brain damage.
Initially, small muscle twitches and constant movements of the limbs are observed. Later, complete chorea develops. It first affects the hands and head, the person makes various grimaces and opens his mouth. Then the lower limbs are affected. These involuntary movements increase significantly during stress, but at the same time do not appear at all during sleep. The movements progress, the person becomes incapable of independence. Signs appear on both sides of the body.
A typical symptom of Huntington's chorea is a gait disorder that sometimes resembles dancing. At a later stage, falls and uncertain movements occur. Involuntary movements may no longer appear, rigidity develops, and it is difficult for the person to move.
The disease is often accompanied by speech impairment (it becomes incomprehensible). In addition, there is a swallowing disorder, leading to serious problems with eating. Eating disorders cause weight loss.
Most cases of Huntington's disease involve urinary incontinence.
Significant manifestations of the genetic disease Huntington's chorea are mental problems. Initially there is minor irritability, then other signs appear, such as:
- aggressiveness;
- apathy;
- ruthless behavior;
- tendency to steal;
- decreased interest in appearance;
- general personality changes.
Although others notice these problems, few attribute them to illness.
Depression occurs already in the early stages of the disease. There is fear of a new day, fear of loss of productivity, failure, lack of energy, and the will to act. A person becomes indifferent to the world around him, gets irritated, has difficulty feeling joy, does not concentrate, and suffers from memory impairment. As a result of mental problems, loss of appetite, weight loss, and sleep disturbances occur. The patient may even have thoughts of suicide. Part of depression is anxiety disorder.
Depression may alternate with a period of hypomania, which is characterized by a feeling of fullness of energy and activity.
The patient lacks insight and self-criticism, so it is difficult for him to understand that he can no longer continue to make decisions about supporting his family or driving a car.
Serious problems include hallucinations and delusions. Hallucination is a perceptual disorder in which the patient cannot distinguish between reality and the pictures that his imagination draws. He perceives auditory and visual hallucinations as true, attempts to change his mind are pointless. Delusion is a thinking disorder in which patients suffer from persecution mania, believe that they have a special origin and abilities, immortality, impunity, etc.
The main clinical symptoms of Huntington's disease include dementia, which occurs in all patients. The person stops doing normal work, cannot concentrate, and constantly forgets.
Reduced mobility and dementia are some of the saddest manifestations because they deprive a person of independence for life.
There are 3 main types of Huntington's disease:
- Juvenile form. Appears by age 20. With this type, involuntary movements are usually absent, but muscle stiffness occurs. There is a rapid development of dementia, personality disorders, and speech disorders.
- Classic shape. It appears at the age of 35-50 years and occurs in 90% of cases. Characterized by the typical course described above.
- Late start form. Appears after 60 years. Of all three types, this is one of the mildest - its course is slow, there is no significant dementia, the person retains independence.
Causes of chorea
The pathogen that causes chorea is still unknown, but many authors indicate a connection between chorea and rheumatism.
The reason for this is the frequent coincidence of these two diseases, as well as the presence in the heart of patients with chorea of changes in Ashof-Talalaev nodules characteristic of rheumatism. Some authors consider chorea as one of the many manifestations of rheumatic infection, as a special form of rheumatism, and speak of chorea as rheumatism of the brain. However, one cannot ignore the fact that chorea can also occur after scarlet fever, tonsillitis, diphtheria and even malaria. It has even been suggested that chorea is related to tuberculosis infection.
Chorea affects children aged 5 to 15 years, most often at 10-12 years of age, and girls are much more likely than boys. At the age of 17-25 years, chorea affects almost exclusively women.
Sometimes outbreaks of chorea observed in children's groups arise as a result of imitation of a sick person, which children with an unbalanced nervous system are prone to. Isolation of the sick person immediately stops the “epidemic”.
Chorea most often occurs in autumn and spring, that is, during the period of greatest prevalence of rheumatism and influenza.
Anatomical changes in chorea have not been sufficiently studied, which is explained by the extremely low mortality rate in this disease. The cause of death is most often heart damage. Nodules characteristic of rheumatic fever are localized not only in the heart, but also in the striatum of the brain. They represent inflammatory changes in the area of the smallest vessels, expressed in infiltration of the perivascular spaces and pinpoint hemorrhages.
The development of chorea is usually preceded by some kind of infectious disease, most often rheumatism.
Diagnostics
Fundamental testing is genetic, demonstrating multiplication of CAG triplets. Based on the level of mutations, it determines the approximate age at which the disorder may appear. Such testing requires very strict ethical rules. Only patients who consider it appropriate and who fully agree with the test can be tested. It is completely unacceptable to force a person to undergo testing or to convince him otherwise. Due to the impossibility of treatment, it is necessary to link these tests with the support of a psychologist and, if necessary, social workers.
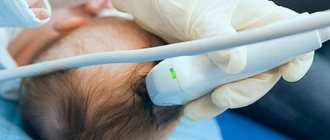
As ancillary studies (as well as as part of differential diagnosis), CT or MRI can be performed. The purpose of these examinations is to determine if there is any disorder in the basal ganglia, especially the caudate nucleus.
A special test is performed in patients suspected of having Huntington's disease. It confirms or excludes suspicion with 100% certainty. The patient's signature is always required to document familiarization with the diagnostic rules.
A predictive (presymptomatic and prenatal) test is performed on asymptomatic people with a family history of the disease who want to know if they are affected by the disease. But a genetic test only confirms the presence of a mutation and prerequisites for the disease, and not the diagnosis itself.
During pregnancy, in women who develop Huntington's chorea, diagnosis may involve a test to determine the genetic status of their unborn child. This prenatal test is performed by collecting amniotic fluid (amniocentesis).
Individuals seeking presymptomatic testing should carefully consider the implications. They have the right not to know their genetic status.
Due to liability concerns, pre-symptomatic testing is not available for individuals under 18 years of age.
Diagnosis of Huntington's chorea
When diagnosing Huntington's chorea, the doctor primarily relies on family history, physical examination, and analysis of the patient's neurological condition. If suspected, laboratory and instrumental examinations are prescribed, without which a diagnosis cannot be made.
The main types of examinations for diagnosing Huntington's chorea:
- genetic research, DNA test;
- electroencephalography;
- computed tomography (CT) of the brain;
- magnetic resonance imaging (MRI);
- positron emission tomography (PET);
- blood chemistry;
- intrauterine genetic testing of the fetus.
Treatment
Huntington's disease is currently incurable. Doctors are trying to counteract many of its manifestations. The disease requires the cooperation of specialists from the fields of neurology, psychology, psychiatry, physiotherapy, occupational therapy, speech therapy, etc.
In very serious cases of involuntary movements, antipsychotics, sedatives or antipsychotics are used. They are also used in cases of delusions, hallucinations, aggression, and anxiety. However, such drugs have many side effects, so they are used only in really severe cases, after a careful assessment of the possible risks.
During the treatment of Huntington's chorea, it is important to combat weight loss by eating high-calorie foods (more than 5000 kcal/day). It is advisable to discuss nutrition with a nutritionist. It is recommended to eat fatty meats, whole dairy products, fatty fish, sweet juices, potatoes, and chocolate.
Antidepressants are effective against depression. For sleep disturbances caused by anxiety, sleeping pills are taken on the recommendation of a doctor. It is also possible to use traditional methods, for example, soothing herbal teas.
Unfortunately, the most serious manifestation of the disease – dementia – is completely incurable. You can't even slow him down.
Complications
One of the complications is infection. The most common infection is pneumonia, which can be fatal to the patient due to weakened immunity.
Vascular disorders and blockage of arteries can lead to heart failure. A serious complication is difficulty swallowing and eating. The patient is losing a lot of weight and is exhausted.
All other complications are related to symptoms related to the disease itself. Changes in behavior are especially disruptive. Often a person becomes self-centered, aggressive, and obsessive. A complication of this behavior is a higher propensity for addiction. The danger is posed by suicide tendencies, which in Huntington's disease are much higher than the average population.
Consequences
Illness in elderly people
A few years after the onset of the disease, a person loses the ability to self-care and becomes disabled. Losing control over your own body leads to irreversible consequences.
Often, during a fall, patients receive serious injuries; when eating, suffocation may occur due to involuntary muscle contraction.
Mental disorders are accompanied by obsessive thoughts and suicidal tendencies.
TEMPERAMENT
It is important to know
Huntington's disease is associated not only with the patient himself, but also with his family and environment. The biggest concerns relate to the health of the offspring - the genetic risk is very high. It is also not easy to explain to children what is really going on with their parents. Why does mom behave so strangely, what she does with her hands, etc. Often the topic of the disease becomes taboo in the wider family or society, which aggravates the patient’s mental state.
The life of the partner of a sick person is turned inside out. He gradually loses his free time, which he should devote to caring for him. Most people living in a family with a sick person develop depression, a negative attitude towards the world, outbursts of anger, constant sadness, etc. Cases of addiction to alcohol are common.
Unfortunately, in our country there is no specialized institution for long-term or short-term treatment of this disease. At the same time, social support plays a vital role in helping patients and their families.