Нервно-мышечные заболевания
Это группа наследственных заболеваний, которые передаются на генетическом уровне (Х сцепленные, аутосомно-доминантные, аутосомно–рецессивные формы, формы наследуемые по материнской линии-митохондриальные). Около трети случаев заболеваний является результатом новых мутаций. С учетом структурной организации выделяют следующие уровни поражения нервно-мышечной системы:
- Первично-мышечные поражения (мышечная дистрофия Дюшена, Беккера, миопатии, миотонии).
- Синаптические поражения (нарушения нервно-мышечной передачи- миастении).
- Нейрональные поражения (спинальная мышечная атрофия).
Что важно знать при врождённых мышечных дистрофиях / миопатиях?
- Семейное руководство по медицинскому уходу при врожденной миопатии (если руководство не отображается во врезке ниже, то вы можете его просмотреть по ссылке https://goo.gl/woCsw6):
- Семейное руководство по медицинскому уходу при врожденной мышечной дистрофии (если руководство не отображается во врезке ниже, то вы можете его просмотреть по ссылке https://goo.gl/YKGySX
- Для врачей «Заявление на основе консенсуса касательно стандартного лечения врожденных миопатий» (если руководство не отображается во врезке ниже, то вы можете его просмотреть по ссылке https://goo.gl/sV4B1B):
СИМПТОМЫ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ
Основной признак нервно-мышечных заболеваний – это слабость мускулатуры. Клиническая картина зависит от области поражения (плечевой пояс, бедра, таз, нижние конечности, мышцы лица, дыхательная мускулатура).В зависимости от формы заболевания за мышечной слабостью и повышенной утомляемостью при физических нагрузках развиваются мышечные атрофии / псевдогипертрофии, появляются фасцикуляции, угнетаются сухожильные рефлексы, нарушается походка, появляются особые двигательные приемы при вставании из положения лежа и сидя. Нервно-мышечные заболевания протекают по стадиям. Определить этап развития патологических процессов поможет врач невролог при помощи медицинской диагностики.
Наследственные пароксизмальные миоплегии
Пароксизмальные миоплегии – редко встречающиеся наследственные заболевания у детей. Характеризуется периодическими приступами паралича мышц скелета. Наследуется по принципу, при котором для проявления болезни достаточно одного мутантного аллеля, локализованного в аутосоме.
Выделяют три формы:
- гиперкалиемическая;
- гипокалиемическая;
- нормокалиемическая.
Гипокалиемической формой пароксизмальной миоплегии чаще всего болеют мужчины.
Первые приступы заболевания появляются с 10 лет.
Приступ возникает в утренние или ночные часы. При этом чувствуется слабость в конечностях, шее, часто доходящая до паралича. В отдельных случаях паралич распространяется на лицевую мускулатуру и дыхательные пути.
Приступ сопровождается жаждой гиперемией, потливостью. Длительность может составлять от часа до семи дней. Женщины обычно испытывают приступ в первый день менструаций.
Гиперкалиемическую форму можно встретить реже, ее приступы начинаются в младшем возрасте. Приступ провоцируют холод и длительное бездействие мышц. Он начинается с расстройства чувствительности лицевых мышц, рук и ног.
Третья форма болезни проявляется до 10-летнего возраста. Приступы проходят в течение нескольких дней или недель. Они могут быть спровоцированы низкой температурой окружающей среды, употреблением алкоголя, большими физическими нагрузками.
ДИАГНОСТИКА
Диагностическая задача состоит в установлении уровня поражения нервно-мышечного аппарата, установить причину и определить есть ли специфическое лечение.
- Электронейромиография и электромиография. Диагностические методы помогают определить первичную и вторичную миопатию. Оценить также степень поражения спинного мозга и периферических нервов.
- Лабораторные тесты. Обязательно сдается клинический и биохимический анализ крови, чтобы определить уровень электролитов, белка, лактата, КФК, АлАТ, АсАТ, миоглобина а также гормонов щитовидной железы.
- Мышечная биопсия с патоморфологическим исследованием мышечной ткани. Результаты диагностики помогают установить точный диагноз (миодистрофия Дюшена, Беккера), что важно для подбора правильной терапии.
- Молекулярно-генетическая диагностика — максимально информативный метод диагностики, который назначается при нервно-мышечных заболеваниях.
- Магнитно-резонансная томография головного мозга и шейного отдела позвоночника.
- Электрокардиограмма (ЭКГ) оценивает сердечный ритм.
- ЭХО КГ выявляет гипертрофическую, дилатационную кардиомиопатию.
Миотонии
Миотония – это замедленное расслабление мышцы после ее напряжения.
Болезнь разделяют на три вида:
- миотония действия;
- перкуссионная миотония;
- электромиографическая миотония.
Если больной с миотонией действия сожмет пальцы в кулак, а затем попытается быстро их разогнуть, то ему понадобится время, чтобы полностью выпрямить ладонь.
Вторая разновидность отличается сокращением мышц при интенсивном постукивании молоточком.
Чтобы обнаружить электромиографическую миотонию, потребуется игольчатый разряд. При этом аппарат показывает разряды высокой частоты, которые сначала учащенные, а потом снижают частоту.
ЛЕЧЕНИЕ НЕРВНО-МЫШЕЧНЫХ ЗАБОЛЕВАНИЙ
Что касается лечения, то специфической терапии не существует. Терапевтический подход основывается на создании предпосылок к успешной реабилитации и обеспечении высокого качества общего и специализированного ухода.
- Медикаментозная терапия. Цель – компенсация энергетического дефицита мышечной ткани, улучшение тканевого метаболизма и периферического кровообращения. Применяются миотропные препараты, анаболические стероиды. В случае миодистофии Дюшена применяются глюкокортикостероиды. В случае миастении применяются антихолинэстеразные препараты. При вторичных повреждениях и / или дисфункции центральной нервной системы проводятся курсы нейропротективной и нейротрофической терапии. Медикаментозная терапия бульбарных, псевдобульбарных расстройств.
- Физиотерапия. Цель – коррекция метаболических нарушений, улучшение нервно-мышечной проводимости, нормализации мышечного тонуса, профилактика развития контрактур.
- Лечебная физкультура. Цель – предупреждение развития и коррекция контрактур конечностей, поддержание двигательной активности, улучшение мышечного тонуса, задержка развития атрофий, предупреждение осложнений, вызванных малоподвижностью. Обучение пациента пользоваться приспособлениями, позволяющими сидеть и стоять с поддержкой.
- Ортопедическая коррекция.
- Раннее выявление нарушения глотания (дисфагии) и возможности медикаментозной коррекции (совместно с ЛОР врачами, по результатам назофарингоскопии — функциональное состояние надгортанника, наличие регургитации, а у детей старшего возраста + анализ результатов рентген контрастного исследования акта глотания).
- Неврологическая оценка дыхательной функции (анализ ЭНМГ диафрагмальных нервов + УЗИ диафрагмы, анализ ФВД, пульсоксиметрии и капнометрии, механики и типе дыхания, данные ЭХО КГ, ЭКГ с заключением кардиолога).
Неврологическое обследование пациентов на предмет дыхательной недостаточности позволяет уточнить ее причины, обратимость нарушений и степень их компенсации. В случае сформировавшейся зависимости от искусственной вентиляции легких работа в команде врачей с созданием многофункциональной терапевтической программы, которая направлена на улучшение физического состояния и восстановление нормальных жизненных функций. выбор методов лечения и их очередности (медикаментозные и не медикаментозные, включая управление параметрами респираторной поддержки, дыхательную гимнастику, лечебную физкультуру, физиотерапевтические методики, психологическую помощь и прочее).
Миастения
Миастения — заболевание, вызывающее нарушение нервномышечной передачи и проявляющееся слабостью и патологической утомляемостью скелетных мышц.
Этиология и патогенез.
Приобретенная миастения связана с образованием антител против ацетилхолиновых рецепторов постсинаптической мембраны нервно-мышечного синапса, которые блокируют передачу возбуждения с нервов на мышцы. В патогенезе аутоиммунной реакции активную роль, по-видимому, играет вилочковая железа (тимус), однако причины ее развития остаются неясными. Значительно более редкая врожденная миастения обусловлена генетически детерминированным дефектом нервно-мышечных синапсов. Неонатальная миастения — преходящее состояние, наблюдающееся у младенцев, родившихся от матерей, страдающих миастенией, и обусловленное переходом через плаценту материнских антител к ацетилхолиновым рецепторам.
Клиническая картина.
Миастения может возникнуть в любом возрасте, но наиболее высокая заболеваемость отмечается в двух возрастных категориях: от 20 до 40 и от 65 до 75 лет. В подавляющем большинстве случаев заболевание первично вовлекает глазные мышцы, поэтому вначале больные жалуются на эпизодическое опущение века и двоение в глазах. В последующие 1-2 года у большинства больных в процесс вовлекаются мимические и бульбарные мышцы, мышцы шеи, конечностей и туловища с развитием генерализованной формы заболевания. Но у части больных заболевание не распространяется далее наружных мышц глаза (глазная форма). Характерны выраженные колебания симптоматики в течение суток, в связи с этим болезнь нередко принимают за истерию. Феномен патологической мышечной утомляемости проявляется нарастанием симптоматики на фоне физической нагрузки (например, усиление слабости жевательных мышц во время еды, ослабление голоса во время беседы). После отдыха симптомы уменьшаются. Характерно отсутствие вегетативных нарушений (нарушений иннервации зрачка или тазовых расстройств), мышечных атрофий, снижения сухожильных рефлексов, нарушений чувствительности. При неврологическом осмотре выявляется снижение силы, нарастающее при повторении движений. Для выявления патологической утомляемости мышцы, поднимающей верхнее веко, больному предлагают фиксировать взор, отведя глаза вверх, для выявления слабости мышц плечевого пояса — поднять руки вверх на 30-60 с, для обнаружения утомляемости мышц гортани — сосчитать вслух до 100. Характерно избирательное вовлечение мышц (например, сгибатели шеи оказываются более слабыми, чем разгибатели), что позволяет отличить миастению от астении или истерии. У больных с генерализованной миастенией иногда возникает быстрое ухудшение состояния с развитием дыхательной недостаточности, связанной со слабостью дыхательных мышц или бульбарной мускулатуры (миастенический криз). Криз может возникнуть вследствие неблагоприятного течения заболевания (иногда он бывает первым проявлением миастении), на фоне инфекции, электролитных нарушений (гипокалиемия, гипермагнезиемия) или приема препаратов, нарушающих нервно-мышечную передачу. Тяжелая дыхательная недостаточность при кризе может развиться очень быстро, в течение нескольких минут. О ее приближении свидетельствуют одышка, неспособность сглатывать слюну и держать голову прямо, ослабление голоса. Реже нарастание мышечной слабости и дыхательной недостаточности бывает вызвано передозировкой антихолинергических средств (холинергический криз). В пользу этого варианта криза свидетельствуют в основном вегетативные нарушения, связанные с активацией ацетилхолиновых рецепторов: узкие зрачки и парез аккомодации, гиперсекреция слюны и бронхиальной слизи, кишечные колики, понос, рвота, брадикардия, а также генерализованные мышечные подергивания. Но у части больных клинически отдифференцировать миастенический криз от холинергического бывает практически невозможно.
Диагностика.
Для подтверждения диагноза миастении проводят прозериновую пробу с 2 мл 0,5 % раствора прозерина, который вводят подкожно, и наблюдают за эффектом в течение 40 мин. У больных с миастенией при этом наблюдается значительное уменьшение, а иногда и полное исчезновение симптомов болезни. Для коррекции возможного побочного действия прозерина: брадикардии, бронхоспазма, артериальной гипотензии — следует также иметь наготове шприц с 0,5- 1 мл 0.1 % раствора атропина и мешок Амбу. При введении прозерина возможны и другие нежелательные эффекты — гиперсаливация, слезотечение, мышечные подергивания, диарея, кишечная колика, тошнота, недержание мочи и кала. Диагноз подтверждают также с помощью электромиографии, определения содержания антител к ацетилхолиновым рецепторам. У взрослых с подтвержденным диагнозом миастении показана компьютерная томография грудной клетки для исключения опухоли или гиперплазии вилочковой железы, выявляющихся у значительной части больных.
Лечение.
Для уменьшения слабости и патологической утомляемости мышц прежде всего применяют анти холин эстеразные средства, тормозящие распад ацетилхолина в синапсе,- пиридостигмин (калимин) и неостигмин (прозерин). Действие калимина начинается через 30-60 мин после приема препарата и продолжается 3-6 ч. Лечение начинают с 30 мг 3 раза в день, затем дозу повышают до 60-120 мг 4 раз в день. Хотя пиридостигмин эффективен у большинства больных, но лишь у небольшой части из них симптомы регрессируют полностью. Действие препарата на различные мышцы бывает неодинаковым: в отношении одних его доза может быть недостаточной, в отношении других — избыточной. Дальнейшее наращивание дозы может усиливать слабость в последнем случае. Чтобы избежать передозировки, очередную дозу следует принимать не ранее, чем появятся признаки окончания действия предыдущей дозы. Частыми побочными эффектами являются боли в животе, тошнота, диарея, гиперсаливация. Иногда для их уменьшения назначают атропин (0,5 мг внутрь), однако регулярный его прием невозможен из- за его токсического действия (тем не менее больным полезно иметь атропин при себе). Побочные реакции можно уменьшить, если снизить разовую дозу антихолинэстеразных средств, увеличив частоту его приема, или принимать препарат во время еды. Прозерин обладает более короткой продолжительностью действия. Часто его назначают внутрь (15-30 мг) или парентерально (0,5-1,5 мг) для получения кратковременного дополнительного эффекта, например, перед обедом. Одновременно больным часто назначают препараты калия. При недостаточной эффективности антихолинэстеразных средств назначают кортикостероиды. Они вызывают улучшение у 70 % больных, но в первые 3 нед, особенно если лечение начато с высокой дозы, слабость мышц (в том числе бульбарных и дыхательных) может нарастать. При достижении стойкого эффекта приступают к медленному снижению дозы. В значительной части случаев больные вынуждены принимать поддерживающую дозу препарата на протяжении многих лет. В тяжелых случаях при плохой переносимости кортикостероидов назначают иммуносупрессоры (азатиоприн, реже циклоспорин и циклофосфамид). Удаление вилочковой железы (тимэктомия) показано больным до 60 лет с генерализованной формой заболевания, а также при наличии опухоли вилочковой железы (тимомы). Осуществляя уход за больным миастенией, медицинская сестра должна оказывать им помощь в соблюдении гигиенического режима, в питании (особая осторожность необходима при нарушении глотания), наблюдать за состоянием двигательных и дыхательных функций. Выдавать лекарственные средства следует строго по назначению врача. Целый ряд лекарственных препаратов способен усилить симптомы миастении, в том числе некоторые антибактериальные средства (стрептомицин, гентамицин и другие аминогликозиды, тетрациклин, ампициллин, эритромицин, ципрофлоксацин, клиндамицин, сульфаниламиды), бета-блокаторы, лидокаин, хинин, новокаинамид, антагонисты кальция, антиэпилептические средства (дифенин, карбамазепин, барбитураты), аминазин, амитриптилин, диазепам (реланиум) и другие бензодиазепины, миорелаксанты, диуретики (за исключением калий сберегающих), соли магния и др. При появлении признаков миастенического криза больной должен быть экстренно госпитализирован в отделение интенсивной терапии. Транспортировку лучше производить в положении полусидя. Во время транспортировки прежде всего следует позаботиться о проходимости дыхательных путей и предупреждении аспирации; необходимо удалить слизь из глотки, дать кислород (через маску или назальный катетер). Иногда показана интубация. В отсутствие признаков передозировки антихолинэстеразных средств (!) можно ввести подкожно 1 — 2 мл 0,05 % раствора прозерина. Внутривенное введение препарата дает более быстрый эффект, но чревато опасностью остановки сердца, поэтому к нему прибегают лишь в наиболее тяжелых случаях. Предварительно вводят внутривенно или подкожно 0,5 мл 0,1 % раствора атропина. Дальнейшее введение прозерина возможно только при получении положительного результата от первого введения. В условиях отделения интенсивной терапии налаживают регулярный контроль за состоянием дыхательной функции и проходимостью дыхательных путей. При развитии дыхательной недостаточности проводят интубацию и приступают к искусственной вентиляции легких. Проводят коррекцию электролитных нарушений. При признаках инфекции назначают антибиотики (предпочтительнее циклоспорины). Больные часто бывают возбуждены, но вводить седативные препараты, как правило, не следует, так как многие из них усугубляют мышечную слабость. Ободряющие слова и деловитость персонала часто в достаточной степени успокаивают больного, в тяжелых же случаях вводят галоперидол (1 мл 0,5 % раствора внутривенно или внутримышечно). Наилучшие результаты при кризе дает плазмаферез. Иногда при кризе используют также кортикостероиды (например, преднизолон, до 100 мг/сут внутрь), но при этом возможно первоначальное нарастание слабости и дыхательной недостаточности. Если же холинергический компонент криза надежно исключен, то в отсутствие потребности в искусственной вентиляции легких продолжают введение прозерина. С началом искусственной вентиляции легких, которую обычно проводят 3-6 дней, прозерин отменяют либо его дозу снижают вдвое. При холинергическом кризе антихолинэстеразные препараты временно отменяют, восстанавливают проходимость дыхательных путей, подкожно вводят атропин (по 0,5-1 мл 0,1 % раствора каждые 2 ч) до появления сухости во рту, назначают реактиваторы холинэстеразы, при необходимости прибегают к интубации и искусственной вентиляции легких.
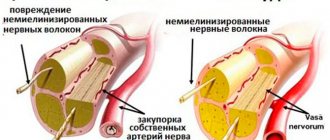
ПД распространяется посредством активации Na + -каналов до нервных окончаний, где он деполяризует клеточную мембрану, что приводит к открытию потенциалзависимых Ca 2+ -каналов. Ионы Ca 2+ , входящие в нервные окончания, запускают выход из пресинаптической мембраны везикул, содержащих АХ, вследствие чего последний высвобождается в синаптическую щель. Затем АХ связывается с рецепторами на субсинаптической мембране и открывает неспецифические катионные каналы. Деполяризация субсинаптической мембраны распространяется на постсинаптическую мембрану, где после открытия потенциалзависимых Na + -каналов возникает ПД, который быстро распространяется по всей мембране мышцы. АХ разрушается с помощью ацетилхолинэстеразы, образовавшийся холин вновь захватывается нервным окончанием и повторно используется для синтеза АХ.
Патологические изменения могут затрагивать любой элемент этого процесса. Местные анестетики, например, ингибируют потенциалзависимые Na + -каналы нейронов, нарушая таким образом нервную передачу к концевой пластинке нервно-мышечного синапса. Ca 2+ -каналы могут быть блокированы антителами. Ботулотоксин инактивирует белок синаптобревин, который отвечает за связывание везикул, содержащих АХ, с плазматической мембраной, т. е. за высвобождение АХ Ацетилхолиновые рецепторы, так же как и Ca 2+ -каналы, могут блокироваться антителами, которые, кроме того, ускоряют интернализацию и разрушение этих рецепторов. Рецепторы могут быть блокированы и кураре, которое, не обладая своим собственным эффектом, конкурентно ингибирует связывание АХ с рецепторами.
Сукцинилхолин (суксаметония хлорид) приводит к продолжительной стимуляции рецепторов, длительной деполяризации постсинаптической мембраны, вызывая тем самым инактивацию постсинаптических Na + -каналов. Благодаря подобному действию он способен, как и кураре, блокировать нервно-мышечную передачу импульсов. В низких концентрациях вещества, ингибирующие ацетилхолинэстеразу (например, физостигмин), облегчают неровно-мышечную передачу путем увеличения доступности АХ в синаптической щели. Однако в высоких дозах они замедляют нервно-мышечную передачу, т. к. высокие концентрации АХ и сукцинилхолина вызывают продолжительную деполяризацию субсинаптической мембраны, инактивируя тем самым постсинаптические Na + -каналы. Повторный захват холина нервными окончаниями может подавлять ионы Mg 2+ и гемихолин.
Важнейшим заболеванием, при котором поражаются концевые пластинки нервно-мышечных синапсов, является myasthenia gravis, характеризующаяся параличом мышц из-за блокады нервно-мышечной передачи импульсов. Это заболевание обусловлено образованием антител к рецепторам АХ на субсинаптической мембране, ускоряющих разрушение этих рецепторов. Это аутоиммунное заболевание могут провоцировать вирусные инфекции, при которых происходит стимуляция экспрессии молекул МНС, что облегчает распознавание антигена иммунной системой. Миастения может встречаться также у пациентов с доброкачественной опухолью тимуса. Образование таких аутоантител чаще имеет место у лиц — носителей специфических подтипов (DR3 и DQw 2) МНС класса II или HLA. В редких случаях миастения вызвана генетическими дефектами каналов, рецепторов АХ или ацетилхолинэстеразы. У пациентов, страдающих myasthenia gravis, повторная стимуляция двигательных нервов сначала будет вызывать образование в мышцах нормальных суммированных ПД, амплитуда которых, однако, будет уменьшаться вследствие прогрессирующего нарастания «усталости» нервно-мышечной передачи.
Другим иммунным аутоагрессивным заболеванием, при котором нарушается нервно-мышечная передача, является синдром псевдомиастении Ламберта-Итона. Это состояние часто развивается у больных мелкоклеточным раком легких. Ca 2+ -каналы в плазматической мембране опухолевых клеток сенсибилизируют иммунную систему и стимулируют образование антител, которые взаимодействуют также с Са2,-каналами концевых пластинок нервно-мышечных синапсов. Благодаря ингибированию Ca 2+ -каналов суммированный ПД мышц сначала маленький, но затем он постепенно нормализуется, т. к. повторная стимуляция увеличивает количество Ca 2+ накапливающегося в нервных окончаниях.
Ацетилхолин секретируется двигательными нервными окончаниями не только при возбуждении, но и в покое. Различие состоит лишь в том, что в покое выделяются малые порции — «кванты» — ацетилхолина, а под влиянием нервного импульса в синаптическую щель одновременно выбрасывается значительное количество таких «квантов». «Квант» представляет собой «пакет» молекул медиатора в единичном пузырьке нервного окончания, изливающем свое содержимое в синаптическую щель. В концевой пластинке различных животных в каждом «кванте» содержится до 2000 молекул ацетилхолина. Выделение отдельных квантов в синаптическую щель в состоянии покоя вызывает кратковременную слабую деполяризацию постсинаптической мембраны мышечного волокна. Такая деполяризация получила название миниатюрного потенциала, поскольку она по своей амплитуде (0,5 мВ) в 50-80 раз меньше ПКП, вызываемого одиночным нервным импульсом. Миниатюрные потенциалы возникают обычно с частотой примерно один в секунду, они зарегистрированы не только в нервно-мышечных соединениях, но и в синапсах нервных клеток ЦНС.
Врождённые не прогрессирующие миопатии
В большинстве случаев поражается область нижних конечностей, реже – верхних, в исключительных случаях встречается поражение краниальной мускулатуры – нарушение мимики, движения глаз.
В процессе развития и роста ребёнка отмечаются проблемы с моторикой, дети часто падают, поздно начинают сидеть и ходить, не могут бегать и прыгать. При этом нет нарушений интеллекта. К сожалению, этот тип миопатий неизлечим.

Миопатия
Термин миопатия (миодистрофия) объединяет достаточно большую группу заболеваний, которые объединены общим признаком: первичным поражением мышечной ткани. Развитие миопатии могут спровоцировать различные факторы: наследственность, вирусное поражение, нарушение обмена веществ и ряд других.
К воспалительным миопатиям (миозитам) относятся заболевания, вызванные воспалительным процессом. Они развиваются в результате аутоиммунных нарушений и могут сопровождаться другими заболеваниями аналогичной природы. Это дерамтомизит, полимиозит, миозит с различными включениями.
Митохондриальные миопатии. Причиной возникновения заболевания являются структурные или биохимические митохондрии. К этому типу заболеваний относятся:
Помимо этих заболеваний, существует ещё ряд редко встречающихся видов миопатий, поражающих центральный стержень, эндокринную систему и т.д.
При активном течении миопатия может привести к инвалидности и дальнейшему обездвиживанию больного.