Hallervorden-Spatz disease (progressive rigidity) is a rare hereditary neurodegenerative disease that affects the basal ganglia and accumulates iron in them.
Cumulative processes occur in the substantia nigra and globus pallidus. Also in the cerebral cortex, changes in nerve cells are observed, the appearance of pigmented neuroglia, and glial cells similar to glia in Alzheimer's disease.
The disease was first described by German researchers Julius Hallervorden and Hugo Spatz, after whom it was named.
However, in modern international medical practice, the disease is called neurodegeneration with brain iron deposition (NBIA). The abandonment of the original name is due to the fact that Julius Hallervorden conducted research on euthanasia in Nazi Germany.
The disease is considered rare, with up to 3 people having the disease per 1 million people. This neurodegeneration can lead to parkinsonism, dystonia, dementia, and death.
Features of genetic failure
Hallervorden-Spatz disease is a hereditary genetic pathology.
The disease can be either familial or sporadic (random), but it is always inherited. The disease belongs to the autosomal recessive type of inheritance. To transmit the disease, both parents must be heterozygous carriers of the disease and have one mutated allele.
The disease is caused by a mutation in the pantothenate kinase gene (PANK2), which is located on chromosome 20 at the 20p12.3-p13 locus. The pantothenate kinase gene is responsible for encoding the protein pantothenate kinase 2, which in turn inhibits the accumulation of the amino acids cysteine and panthetheine.
As a result, chemical compounds with iron ions are formed, which have a negative effect on proteins and trigger peroxidation processes, which leads to programmed cell death of neurons. Instead of dead neurons, glial tissue (auxiliary cells of nervous tissue) grows.
Pathological processes occur in the globus pallidus, red nuclei and substantia nigra (substantia nigra). A greenish-brown substance that contains iron accumulates in them. In addition, spheroid neuroaxonal formations (extensions of axons with proliferation of tubular and membranous structures) are observed in the white matter of the brain, cerebral cortex, peripheral nerve trunks and spinal cord.
Hallervordenspatz disease
The basal ganglia are neural subcortical ganglia located in the central white matter of the hemispheres. They consist of the striatum, striatum and lenticular nucleus. The structure of the latter includes the globus pallidus and the shell. In addition, part of the dark substance, a part of the extrapyramidal system, belongs to the basal ganglia.
Purpose of the basal ganglia:
- regulation of motor and autonomic functions (breathing, cardiovascular system);
- participation in integrative processes of higher nervous activity.
With Hallervorden-Spatz disease, iron accumulates in the globus pallidus and dark substance, which leads to disruption of their function. As a result, motor dysfunctions and mental disorders appear. The disease was first described in 1922 by physicians Julius Hallervorden and Hugo Spatz. It occurs in 3 out of 1,000,000 people.
Circumstances and pathogenesis
Hallervorden-Spatz disease is a hereditary pathology: it is transmitted according to an autosomal recessive principle. But the medical literature describes not only domestic cases of the disease, but also sporadic (random) cases.
The reason for the development of the disease is mutations in the PANK2 gene, located on chromosome 20. It is responsible for encoding pantothenate kinase. This enzyme is involved in the biosynthesis of coenzyme A, which activates the phosphorylation of pantothenate, panthine and N-pantothenylcysteine.
The genetic deficiency leads to decreased production of pantothenate kinase. As a result, the amino acid cysteine accumulates in the basal ganglia, which has the property of binding iron ions. As a result, stable compounds are formed in the brain, provoking the destruction of neuronal proteins.
Apoptosis (death) of neurons is improved by oxidative reactions involving free radicals. As a result, the destroyed neurons are replaced by glia - auxiliary cells of the nervous tissue.
The pathological picture of Hallervorden-Spatz disease is characterized by such indicators as:
- accumulation of iron-containing pigment in the globus pallidus, dark substance and red nuclei, why these areas take on a yellowish-brown hue;
- neuroaxonal formations (axons with local extensions) in the basal ganglia, cerebral cortex and peripheral nerves;
- death of neurons in various parts of the brain;
- diffuse demyelination - destruction of the sheath of nerve fibers.
At the same time, non-specialized iron metabolism is not disturbed - its amount in the blood serum does not exceed the norm.
Forecast
Hallervorden-Spatz disease is characterized by steady progression of symptoms. The most aggressive course is characteristic of the childhood form: 6-15 years after the end of the manifestation of the pathology, complete disability occurs.
The late version of the disease has a more favorable prognosis, especially if it is accompanied by mild dementia. Thanks to therapy, the degree of symptoms can be reduced and the patient’s ability to self-care maintained. The average duration of the atypical form of the disease is 20 years or more.
Features of the clinical picture
Signs of this pathology may vary in each specific case, and the manifestation of symptoms depends on the form of the disease in the individual.
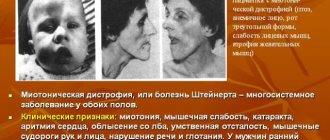
The childhood form of Hallervorden Spatz appears between the ages of 5 and 10 years. It is considered a classic form of the disease.
In the vast majority of cases (90%), the disease begins with torsion dystonia, which affects the leg muscles. The patient experiences muscle contraction, difficulty walking, and changes in gait. Then the muscles of the face, pharynx and trunk are affected.
The presence of blepharospasm, hand spasm, spasmodic torticollis, and facial hemispasm may be present. A third of patients have muscle rigidity, hypokinesia, and tremor (signs of Parkinsonism syndrome).
Typical signs of the childhood form of the disease also include:
- epileptic syndrome;
- aggressiveness;
- mental retardation (occurs due to deterioration of memory and attention);
- asociality;
- visual disturbances (optic atrophy, retinal degeneration);
- mental disorders.
The childhood form of the disease progresses quickly, and complete loss of the ability to move occurs 10-15 years after the onset of the disease.
The teenage form develops and progresses more slowly. At the beginning of the disease the following appear:
- focal torsion dystonia (most often affects the muscles of the limbs and maxillo-oral muscles);
- neuropsychological disorders;
- behavioral disorders;
- intellectual disorders.
The atypical form of the disease is the rarest (15% of cases), and proceeds differently from the first two forms. The most typical symptoms for this form are:
- Parkinsonism syndrome with the inability to maintain balance while standing;
- involuntary movements in different parts of the body, dystonia, chorea and athetosis (slow and jerky movements), hemiballismus (sweeping movements), myoclonus (non-prolonged muscle spasms);
- dementia;
- epileptic syndrome;
- depression, aggression;
- pathological reflexes that arise as a result of destructive processes in the neurons of the brain.
Hallervorden-Spatz disease
Hallervorden-Spatz disease
- a neurodegenerative hereditary pathology caused by iron deposition in the basal ganglia of the brain.
It manifests itself as parkinsonism syndrome, intellectual and mental disorders, hyperkinesis, and visual disorders. The main diagnostic value is the detection of the “eye of the tiger” pattern in the area of the globus pallidus during MRI of cerebral structures.
Treatment is symptomatic: dopamine agonists, valproates, anticonvulsants, neuroleptics, antidepressants. The prognosis is unfavorable.
Hallervorden-Spatz disease was described in 1922 by German morphologists, after whom it received its name. The most typical clinical markers of this pathology include hyperkinesis, parkinsonism syndrome, intellectual decline, optic nerve atrophy, and pigmentary retinopathy.
Hallervorden-Spatz disease is extremely rare. Depending on the time of its manifestation, childhood, juvenile (teenage) and adult forms are distinguished. Previously, the disease was diagnosed only posthumously based on autopsy data.
After the introduction of MRI into practical neurology, intravital diagnosis became possible. A breakthrough in the study of etiology was made in 2001, when it was found that the disease is based on a genetic defect that causes disturbances in the synthesis of the enzyme pantothenate kinase.
After this, Hallervorden-Spatz disease was officially renamed pantothenate kinase-associated neurodegeneration.
Causes of Hallervorden-Spatz disease
Hallervorden-Spatz disease is a genetic pathology that is both familial and sporadic, inherited in an autosomal recessive manner. The genetic substrate is aberrations in the pantothenate kinase gene (locus 20p12.3-p13 of the 20th chromosome). In total, more than 50 mutations are known.
The result of the genetic defect is a decrease in the production of pantothenate kinase, which leads to the accumulation of cysteine in the basal structures. The latter forms stable chemical compounds with iron ions, which adversely affect proteins and trigger the process of peroxidation, leading to neuronal apoptosis.
In place of necrotic neurons, glial tissue grows.
The described pathological process primarily affects the globus pallidus and the substantia nigra (substantia nigra), where extracellular iron deposits with brown pigmentation are morphologically found. In addition, there are spheroid periaxonal formations located in the white cerebral matter, cerebral cortex, spinal cord and peripheral nerve trunks.
The classic variant of Hallervorden-Spatz disease is considered to be an early childhood form with clinical manifestation in the period from 4 to 10 years (usually after 5 years of age). In 90% of cases, the first sign of the disease is torsion dystonia, affecting the leg muscles. The leading complaint is difficulty walking.
Then, as a rule, the process generalizes and spreads to the muscles of the pharynx, face, and torso. Along with generalized variants, multifocal or segmental types of dystonia may be noted. The most commonly observed are writer's cramp, blepharospasm, facial paraspasm, and spasmodic torticollis.
A third of patients show signs of parkinsonism: muscle rigidity and hypokinesia. In some cases, epileptic seizures occur.
Hallervorden-Spatz disease is characterized by cognitive disorders in the form of decreased attentiveness and memory with the gradual development of oligophrenia; mental changes with a predominance of aggressiveness and antisocial behavior. Dysarthria is noted.
Most patients have impaired visual acuity. In 68% of cases they are caused by atrophy of the optic nerves, in 29% of cases by pigmentary retinopathy.
The childhood form of Hallervorden-Spatz disease typically progresses rapidly with complete loss of the ability to move within 10-15 years.
The teenage version of Hallervorden-Spatz disease manifests itself between the ages of 10 and 18 years and is characterized by a slower progression. It debuts with manifestations of focal torsion dystonia, most often in the muscles of the limbs or orthomandibular region. Accompanied by mental, intellectual and behavioral disorders.
The adult form of Hallervorden-Spatz disease debuts after the age of 18. In most patients it occurs in the form of parkinsonism syndrome.
Characterized by impoverishment of active movements (hypokinesia), generalized muscle rigidity (cogwheel symptom), postural tremor and instability (ataxia).
The latter can be assessed using the Tavenard test - attempts to unbalance the patient in a standing position by pushing him forward and to the sides by the shoulders. The main feature is the combination of parkinsonism with various hyperkinesis (myoclonus, focal torsion dystonia, athetosis, hemibalism). The degree of cognitive impairment can vary from complete preservation to progressive dementia. Emotional lability, depression, aggressiveness, and epileptic seizures are possible.
Due to the polymorphism of symptoms, diagnosing Hallervorden-Spatz disease is a difficult task for neurologists.
The main criteria for the disease are the onset before the age of 30, extrapyramidal disorders, steady progression of symptoms, and the presence of a typical MRI picture.
Additional features include the presence of pyramidal signs, progressive intellectual decline, epileptic seizures, optic nerve atrophy, retinal pigmentary atrophy, and autosomal recessive inheritance.
Diagnosis is based on data from neurological status and electroencephalography. If vision is impaired, consult an ophthalmologist, viziometry, and ophthalmoscopy. The type of inheritance is determined by a geneticist by drawing up a family tree. DNA diagnostics is possible (search for mutations in the pantothenate kinase gene).
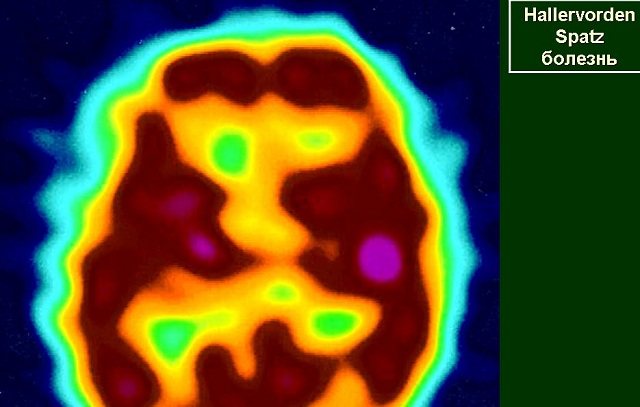
When performing a PET scan of the brain, it is possible to detect reduced metabolism in the pallidum area. The basis for excluding Hallervorden-Spatz disease is the presence of symptoms of another pathology, within the framework of which the existing clinical picture may fit: Wilson's disease, Huntington's chorea, neuroacanthocytosis, Machado-Joseph disease.
The fundamental method for diagnosing Hallervorden-Spatz disease is MRI. In all typical variants of the pathology, a T2-mode MRI of the brain reveals an oval-shaped hyperintense zone located in the globus pallidus region, surrounded by an even larger hypointense zone.
This MRI picture is pathognomonic and is called the “eye of the tiger.” The hypointense zone represents the “eye,” and the hyperintense zone represents the “pupil.” The time at which this sign appears on tomograms is still being debated.
According to some authors, the “eye of the tiger” may appear even before the clinical manifestation of the disease; according to others, it may appear several years after the onset of clinical symptoms.
Treatment and prognosis of Hallervorden-Spatz disease
Currently, there is no effective treatment for Hallervorden-Spatz disease. Attempts at therapy with chelate compounds (deferoxamine) and antioxidants that prevent iron accumulation were unsuccessful.
In this regard, symptomatic treatment is used. Parkinsonism syndrome is an indication for the use of dopamine agonists (piribedil, pramipexole) or amantadine derivatives.
However, with this disease, it is usually resistant to treatment.
For hyperkinesis, valproates and benzodiazepines (diazepam, clonazepam) are used.
For spasticity, muscle relaxants are recommended (baclofen, tolperisone hydrochloride), for epileptic seizures - topiramate or valproate, for cognitive disorders - ipidacrine and choline alfoscerate, for mental disorders - antipsychotics (risperidone, quetiapine, clonazepam), 3rd generation antidepressants (venlafaxine, citalopram, dapoxetine).
Symptomatic therapy for Hallervorden-Spatz disease can reduce the manifestation of clinical symptoms and prolong the patient’s ability to self-care. At the same time, the development of new treatment methods continues. The effectiveness of pantothenic acid is being studied. Data were obtained on the positive effect of magnetic stimulation of the globus pallidus on the course of the disease.
The prognosis depends on the form of the disease. The most unfavorable course is the early form, in which complete disability occurs within 10 to 15 years from the onset of symptoms. The adult option is more favorable, especially in cases where dementia is mild. Its average duration is more than 20 years.
Source: https://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/Hallervorden-Spatz
Symptoms of Hallervorden-Spatz disease
The classic variant of Hallervorden-Spatz disease is considered to be an early childhood form with clinical manifestation in the period from 4 to 10 years (usually after 5 years of age).
In 90% of cases, the first sign of the disease is torsion dystonia, affecting the leg muscles. The leading complaint is difficulty walking. Then, as a rule, the process generalizes and spreads to the muscles of the pharynx, face, and torso. Along with generalized variants, multifocal or segmental types of dystonia may be noted. The most commonly observed are writer's cramp, blepharospasm, facial paraspasm, and spasmodic torticollis.
A third of patients show signs of parkinsonism: muscle rigidity and hypokinesia. In some cases, epileptic seizures occur.
Hallervorden-Spatz disease is characterized by cognitive disorders in the form of decreased attentiveness and memory with the gradual development of oligophrenia; mental changes with a predominance of aggressiveness and antisocial behavior. Dysarthria is noted.
Most patients have impaired visual acuity. In 68% of cases they are caused by atrophy of the optic nerves, in 29% of cases by pigmentary retinopathy.
The childhood form of Hallervorden-Spatz disease typically progresses rapidly with complete loss of the ability to move within 10-15 years.
The teenage version of Hallervorden-Spatz disease manifests itself between the ages of 10 and 18 years and is characterized by a slower progression. It debuts with manifestations of focal torsion dystonia, most often in the muscles of the limbs or orthomandibular region. Accompanied by mental, intellectual and behavioral disorders.
The adult form of Hallervorden-Spatz disease debuts after the age of 18. In most patients it occurs in the form of parkinsonism syndrome.
Characterized by impoverishment of active movements (hypokinesia), generalized muscle rigidity (cogwheel symptom), postural tremor and instability (ataxia).
The latter can be assessed using the Tavenard test - attempts to unbalance the patient in a standing position by pushing him forward and to the sides by the shoulders. The main feature is the combination of parkinsonism with various hyperkinesis (myoclonus, focal torsion dystonia, athetosis, hemibalism). The degree of cognitive impairment can vary from complete preservation to progressive dementia. Emotional lability, depression, aggressiveness, and epileptic seizures are possible.
Diagnosis of Hallervorden-Spatz disease
Due to the polymorphism of symptoms, diagnosing Hallervorden-Spatz disease is a difficult task for neurologists.
The main criteria for the disease are the onset before the age of 30, extrapyramidal disorders, steady progression of symptoms, and the presence of a typical MRI picture.
Additional features include the presence of pyramidal signs, progressive intellectual decline, epileptic seizures, optic nerve atrophy, retinal pigmentary atrophy, and autosomal recessive inheritance.
Diagnosis is based on data from neurological status and electroencephalography. If vision is impaired, consult an ophthalmologist, viziometry, and ophthalmoscopy. The type of inheritance is determined by a geneticist by drawing up a family tree. DNA diagnostics is possible (search for mutations in the pantothenate kinase gene).
When performing a PET scan of the brain, it is possible to detect reduced metabolism in the pallidum area. The basis for excluding Hallervorden-Spatz disease is the presence of symptoms of another pathology, within the framework of which the existing clinical picture may fit: Wilson's disease, Huntington's chorea, neuroacanthocytosis, Machado-Joseph disease.
The fundamental method for diagnosing Hallervorden-Spatz disease is MRI. In all typical variants of the pathology, a T2-mode MRI of the brain reveals an oval-shaped hyperintense zone located in the globus pallidus region, surrounded by an even larger hypointense zone.
This MRI picture is pathognomonic and is called the “eye of the tiger.” The hypointense zone represents the “eye,” and the hyperintense zone represents the “pupil.” The time at which this sign appears on tomograms is still being debated.
According to some authors, the “eye of the tiger” may appear even before the clinical manifestation of the disease; according to others, it may appear several years after the onset of clinical symptoms.
Diagnostic criteria and techniques
Making a diagnosis of Hallervorden-Spatz disease can be problematic for neurologists due to the polymorphism of symptoms.
Main criteria for diagnosis:
- onset of illness before age 30;
- typical picture in MRI results;
- constant development of symptoms;
- extrapyramidal syndromes;
- pyramid signs;
- epileptic seizures;
- cognitive impairment;
- retinal pigmentary degeneration;
- presence of the disease in a family history.
“Eyes of the tiger” on MRI with contrast is a typical picture of Hallervorden Spatz disease.
The disease must be differentiated from other diseases that have a similar clinical picture: Huntington’s chorea, Wilson-Konovalov disease, choreoacanthocytosis, Machado-Joseph disease.
For diagnosis, the patient is referred for the following examinations:
- Study of neurological status by a neurologist, neuropathologist.
- Electroencephalography.
- Consultation with an ophthalmologist . Direct ophthalmoscopy and visual acuity testing are possible.
- Genetic studies to determine the type of inheritance.
- DNA studies (detection of mutations in the PANK2 gene).
- Positron emission tomography of the brain . It is carried out to detect decreased blood circulation in the frontoparietal lobes, striatum, and globus pallidus.
- Magnetic resonance imaging of the brain . Required to detect the "eye of the tiger" - a hyperintense oval area in the hypointense zone that occurs due to iron accumulation. There is no single theory about the development of the “eye of the tiger”; there are assumptions about its occurrence before the onset of clinical manifestations of the disease, and about its appearance years after the onset of the disease. MRI can also detect spheroid neuroaxonal formations.
Treatment
Causal (etiological) therapy is unknown. There have been attempts to treat the enzyme defect. Iron chelators ("traps") such as Deferoxamine have no effect, however, treatment with the iron chelator ferriprox (deferiprone®) has been attempted since 2007. In animal experiments, deep brain stimulation led to increased dystonia and hyperkinesis. Hypokinesia can be treated with levodopa, hyperkinesia with anticholinergics. However, the effect of levodopa in patients with a PANK2 gene mutation is very questionable. Baclofen or benzodiazepines are often prescribed for muscle relaxation and pain relief.
How can modern doctors help?
There are no treatments in modern medicine that can prevent or stop Hallervorden-Spatz disease.
Therapy is aimed at alleviating and relieving the intensity of symptoms:
- For parkinsonism syndrome, dopamine agonists (Pronoran, Pramipexole, Mirapex, Piribedil) and amantadines (Simmetrel, Midantan) are prescribed. However, the syndrome is often resistant to treatment.
- To relieve hyperkinesis, valproates (Konvulex, Depakin, Encorat) and benzodiazepines (Clonazepam, Diazepam) are used.
- To relieve muscle spasticity, muscle relaxants (Mydocalm, Baclofen) are used.
- Epileptic seizures are relieved with valproate , Tomapax.
- For cognitive impairment, Gliatilin and Neuromidin .
- For the treatment of mental disorders, it is recommended to take antipsychotics (Clonazepam, Quetiapine, Rispolent), antidepressants (Dapoxetine, Citalopram, Venlafaxine).
New methods of treating the disease are also emerging. These include therapy by administering pantothenic acid, magnetic stimulation of the brain (globus pallidus).
Sad, but not hopeless
With Hallervorden-Spatz disease, symptoms constantly progress.
The childhood form of the disease is the most severe. Complete disability of an individual occurs 10-15 years from the moment of the first appearance of clinical manifestations. The most favorable development of the disease is predicted in the adult form of the disease. Especially in cases of mild dementia.
Therapy allows the patient to maintain the relative quality of life and his ability to self-care. Life expectancy with the atypical form of Hallervorden-Spatz disease can be more than 20 years.
Symptoms
The leading symptom is unbearable pain in the lumbar region (right or left) with irradiation along the ureter, into the iliac and groin areas, the inner thighs, and external genitalia.
Patients are restless, rush around, cannot find a comfortable position (symptom of a “tiger in a cage”). The cause of pain is a sudden obstruction of one of the ureters due to a stone being pinched in its lumen, or less commonly, blockage of the urethra by a blood clot, pus, etc. The disease occurs in paroxysms. The attack lasts from several minutes to a day or more. When obstructed by a stone, a particularly severe picture develops: nausea, vomiting is observed, at the height of the attack there may be urinary retention, flatulence, and if an infection occurs, the body temperature rises and chills appear. A general urine test reveals hematuria, pyuria, and bacteriuria. The blood test showed leukocytosis, increased ESR. To confirm the diagnosis, pyelography, radiography and ultrasound of the kidneys, ureters, and bladder are done.