Werdnig-Hoffmann disease is a spinal muscular atrophy characterized by degeneration of nerve cells within the lower region of the brain (brain stem) and some motor neurons in the spinal cord (anterior horn).
More than 80% of infants experience severe weakness before the age of 6 months and almost all of them will never achieve the ability to sit independently. Muscle weakness, lack of motor development and poor muscle tone are the main clinical manifestations of this disease. Most babies also have trouble sucking or swallowing. Some show signs of abdominal breathing during the first few months of life. Most children die before age 2 years, but survival may depend on the degree of respiratory function and respiratory support.
Werdnig-Hoffman disease is divided into five subtypes. This division is based on the age at which the first symptoms/manifestations appear, the course and progression of the disease. Subtype 0 is characterized by extreme weakness at birth, and all children require immediate mechanical ventilation almost from birth. These children will never be able to breathe on their own. Subtype 1 is characterized by acute spinal muscular atrophy, with symptoms and signs appearing around 6 months of age. In patients with subtype 2, symptoms and manifestations appear before the age of 1 year. Children with this subtype may learn to sit, but they will never be able to walk. Symptoms and manifestations of subtype 3 (Kugelberg-Welander disease) appear after the age of 1 year. This subtype progresses over a period of time to loss of motor ability. In individuals with the fourth subtype, the first symptoms and manifestations appear at the age of about 10 years.
Werdnig-Hoffman disease. Symptoms and manifestations
The clinical features and rate of progression of Werdnig-Hoffmann disease vary among affected individuals. Early signs of the disease include muscle weakness (often appearing before 6 months of age), decreased muscle tone (hypotonia), abnormal joint flexibility (hypermobility), absent tendon reflexes, and tongue twitching. Most often, children do not gain control over their head; they cannot roll over, sit or stand. Children with this condition may also have difficulty sucking, swallowing, and breathing, have an increased susceptibility to respiratory infections, and often develop other complications that can lead to potentially life-threatening problems within the first few months or years of life.
For children who appear to be developing normally for several months before the first signs of muscle weakness appear, the disorder tends to progress more slowly. As the disease progresses, decreased muscle tone and weakness may begin to gradually spread to almost all muscles except the muscles that control eye movements.
The rate of progression of Werdnig-Hoffmann disease varies. Breathing and bowel problems (constipation) may occur within a few months. The child may completely lose the ability to swallow. Respiratory failure or aspiration of food may also occur in some children. Most children die before age 2 years, but survival may depend on the degree of respiratory function.
Werdnig-Hoffmann amyotrophy
Werdnig-Hoffmann amyotrophy
- This is the most malignant spinal muscular atrophy, developing from birth or in the first 1-1.5 years of a child’s life. It is characterized by increasing diffuse muscle atrophy, accompanied by flaccid paresis, progressing to complete plegia.
As a rule, Werdnig-Hoffmann amyotrophy is combined with bone deformities and congenital developmental anomalies. The diagnostic basis is anamnesis, neurological examination, electrophysiological and tomographic studies, DNA analysis and study of the morphological structure of muscle tissue.
The treatment is weakly effective and is aimed at optimizing the trophism of nervous and muscle tissue.
Werdnig-Hoffmann amyotrophy is the most severe variant of all spinal muscular atrophies (SMA). Its prevalence is at the level of 1 case per 6-10 thousand newborns. The disease has several forms: congenital, intermediate (early childhood) and late.
A number of specialists identify the latter form as an independent nosology - Kugelberg-Welander amyotrophy.
The lack of etiotropic and pathogenetic treatment and early death make the management of patients with Werdnig-Hoffmann disease one of the most difficult tasks facing modern neurology and pediatrics.
Werdnig-Hoffmann amyotrophy
Werdnig-Hoffmann amyotrophy is a hereditary pathology encoded by a breakdown in the genetic apparatus at the level of the 5q13 locus of the 5th chromosome.
The gene in which mutations occur is called the survival motor neuron gene (SMN) - the gene responsible for the survival of motor neurons. 95% of patients with Werdnig-Hoffmann disease have a deletion of the telomeric copy of this gene.
The severity of SMA directly correlates with the length of the deletion site and the concomitant presence of changes (recombination) in the H4F5, NAIP, and GTF2H2 genes.
Every 50th person is a carrier of the altered gene that causes the disease. But thanks to the autosomal recessive type of inheritance, pathology in a child manifests itself only when the corresponding genetic aberration is present in both the mother and father. The probability of having a child with pathology in such a situation is 25%.
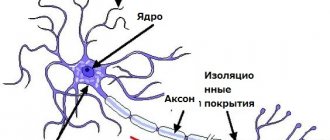
The result of aberration of the SMN gene is the underdevelopment of spinal cord motor neurons localized in its anterior horns.
The consequence is insufficient innervation of the muscles, leading to their severe atrophy with loss of muscle strength and progressive decline in the ability to perform active motor acts.
The main danger is weakness of the chest muscles, without which movements that ensure respiratory function are impossible. At the same time, the sensory sphere remains intact throughout the disease.
SMA I clinically manifests itself before 6 months of age. In utero it may be manifested by sluggish fetal movement. Often, muscle hypotonia is noted from the first days of life and is accompanied by the extinction of deep reflexes.
Children cry weakly, suck poorly, and cannot hold their head up. In some cases (with a later onset of symptoms), the child learns to hold his head up and even sit, but as the disease progresses, these skills quickly disappear.
Characteristic are early bulbar disorders, decreased pharyngeal reflex, and fascicular twitching of the tongue.
This Werdnig-Hoffmann amyotrophy is combined with mental retardation and disorders of the formation of the osteoarticular apparatus: deformities of the chest (funnel-shaped and keeled chest), curvature of the spine (scoliosis), joint contractures. Many patients have other congenital anomalies: hemangiomas, hydrocephalus, clubfoot, hip dysplasia, cryptorchidism, etc.
The course of SMA I is the most malignant, with rapidly increasing immobility and paresis of the respiratory muscles.
The latter causes the development and progression of respiratory failure, which is the main cause of death.
Due to impaired swallowing, food can be refluxed into the respiratory tract with the development of aspiration pneumonia, which can be a deadly complication of spinal amyotrophy.
Early childhood form
SMA II debuts after 6 months of age.
By this period, children have satisfactory physical and neuropsychic development; in accordance with age standards, they acquire the skills to hold their heads, roll over, sit down, and stand.
But in the vast majority of clinical cases, children never have time to learn to walk. Typically, this Werdnig-Hoffmann amyotrophy manifests itself after a child has suffered a foodborne toxic infection or another acute infectious disease.
In the initial period, peripheral paresis occurs in the lower extremities. Then they quickly spread to the upper limbs and muscles of the trunk. Diffuse muscle hypotonia develops, and deep reflexes fade.
Tendon contractures, finger tremor, and involuntary muscle contractions (fasciculations) of the tongue are observed. In the later stages, bulbar symptoms and progressive respiratory failure appear. The course is slower than that of the congenital form of Werdnig-Hoffmann disease.
Patients can live up to 15 years of age.
Kugelberg-Welander amyotrophy
SMA III is the most benign spinal amyotrophy of childhood. Manifests after 2 years, in some cases in the period from 15 to 30 years. There is no mental retardation; patients are able to move independently for a long time. Some of them live to a ripe old age without losing the ability to self-care.
Patients with spinal muscular atrophy type I are under the supervision of pediatric neurologists and neonatologists. The age of manifestation of the disease is of great importance - Werdnig-Hoffmann amyotrophy is characterized by development from birth to 6 months. The history often contains information about late and sluggish fetal movements during pregnancy.
When examining a child, attention is drawn to pronounced muscle hypotonia, the inability to sit independently or hold up the head, and a typical “frog pose” - shoulders raised, arms along the body, legs straightened and turned outward.
Muscle twitching, weakened or absent tendon reflexes, and gross bone deformities (bell-shaped chest, X-shaped lower limbs) are noted.
To confirm the diagnosis, the following additional studies are prescribed:
- Laboratory tests.
A biochemical blood test reveals a slight increase in the concentration of creatine phosphokinase. In some patients, this indicator may be within normal values. Blood gas analysis reveals a decrease in the partial pressure of oxygen (paO2) and an increase in carbon dioxide (paCO2). - Spirometry.
Due to the pronounced muscle weakness of the respiratory muscles, when measuring the function of external respiration, restrictive disorders are noted in the form of a decrease in the vital capacity of the lungs. - ENMG.
When performing needle electroneuromyography, it is possible to record the following changes - a sharp decrease in the conduction velocity and amplitude of evoked potentials, spontaneous bioelectrical activity at rest (fasciculations, fibrillations). - Histology.
A pathomorphological examination of a muscle biopsy reveals fascicular atrophy of muscle fibers, alternating with unchanged muscle tissue, hypertrophied myofibrils and connective tissue growths. - DNA analysis.
A verification test that allows you to reliably establish a diagnosis. The polymerase chain reaction method detects a genetic mutation (deletion) of SMN1 exon 7.
Werdnig-Hoffmann amyotrophy should be differentiated from other genetically determined neuromuscular diseases that have the same rapidly progressive course. These include congenital structural myopathies, juvenile amyotrophic lateral sclerosis, and Fukuyama syndrome.
To undergo treatment, all patients are subject to mandatory hospitalization in a hospital.
In severe situations (for example, with severe hypoxemia due to weakness of the respiratory muscles), patients are transferred to the intensive care unit and connected to a ventilator.
To date, there is no etiotropic therapy for Werdnig-Hoffmann amyotrophy. All measures are symptomatic and palliative in nature. The following treatment methods are used:
- Providing food.
Since the swallowing process is difficult for many patients, special attention is paid to the issue of feeding. The consistency of the food should be semi-solid, the child’s position should be vertical. If necessary, a nasogastric tube is inserted. - Physiotherapy.
To improve metabolism in muscle tissue, sessions of electrophoresis, electrical stimulation with modulated current, and mud applications are performed. - Exercise therapy.
In order to increase muscle tone, regular physical exercises are necessary - passive (performed by a specialist) with a gradual transition to active (performed by the patient himself). - Orthopedic treatment.
To combat osteoarticular deformities, as well as to prevent them, orthopedic devices (corsets, orthoses, immobilizing splints) that fix various parts of the body are used. - Respiratory support.
An important place in treatment is the elimination of oxygen deficiency. Depending on the severity of the patient's condition, oxygen inhalation through a face mask/nasal cannula or non-invasive ventilation through portable ventilators are prescribed.
Drug therapy
To achieve maximum effect, treatment must be comprehensive, carried out continuously and selected individually for a particular patient. Medicines used to treat Werdnig-Hoffmann amyotrophy are as follows:
- Metabolic agents.
To improve metabolic processes in nerve cells and muscle tissue, coenzyme Q10, L-carnitine, and nootropics are prescribed. Also, to stimulate the regeneration of nervous tissue, high doses of B vitamins (B1, B6, B12) are used. - Valproate.
Antiepileptic drugs from the group of valproic acid derivatives are able to increase the formation of survival motor neuron (SMN) protein, which can subsequently improve the clinical course of the disease. - PPIs and prokinetics.
Proton pump inhibitors (pantoprazole) and drugs that accelerate gastrointestinal motility (itopride) help combat gastroesophageal reflux, which often occurs in patients with AVG due to severe swallowing disorders. - Mucolytics and expectorants.
In order to combat respiratory problems such as weak coughing and the accumulation of thick sputum in the respiratory tract, drugs that thin sputum (acetylcysteine) and stimulate expectoration (terpin hydrate) are used.
Surgery
With the development of severe deformities of the chest and spine or extremely pronounced joint contractures, orthopedic surgery is indicated. In bedridden patients suffering from constantly recurrent pneumonia, a tracheostomy is performed. For gastroesophageal reflux that is resistant to drug treatment, laparoscopic Nissen fundoplication is used.
Werdnig-Hoffman disease. Causes
All forms of Werdnig-Hoffmann disease are caused by mutations in the SMN1 gene, located at the 5q11-q13 locus. A second gene, known as SMN2, also plays a role in the development of this disease. The SMN2 gene is located next to the SMN1 gene on chromosome 5. While mutations in the SMN1 gene are implicated in the development of Werdnig-Hoffmann disease, some research groups have obtained evidence that abnormalities in the SMN2 gene may influence the severity of the disease. Individuals with a large number of copies of the SMN2 gene tend to have a mild form of Werdnig-Hoffman disease.
Apparently, the SMN1 and SMN2 genes encode proteins that are necessary for the proper functioning of motor neurons. Some researchers believe that the SMN2 gene produces a partially effective protein necessary for the proper functioning of motor neurons. This is why people with more copies of the SMN2 gene have a mild form of Werdnig-Hoffman disease.
Additional genes may also influence the development of this disease. A greater number of patients with Werdnig-Hoffmann disease have deletions in the NAIP gene. Some researchers have suggested that loss of the NAIP gene and/or various mutations in the SMN genes may play a role in the severity of the disease.
What happens when you get sick
When examining a patient with this pathology, a slight decrease in the volume of the spinal cord is noted. Ganglion cells may atrophy or disappear completely. In the anterior roots, degeneration is detected, a decrease or complete absence of myelination in the nerve fibers, with the deposition of adipose tissue in them.
In the skeletal muscles there are atrophied bundles that intertwine with normal fibers, hyalinosis and a strong proliferation of connective tissue are noted, which replaces muscle tissue.
Werdnig-Hoffmann disease has its own classification. It distinguishes three stages of pathology.
- Congenital, in which symptoms begin to appear in the first few months after birth or immediately after birth.
- Early childhood, in which symptoms are detected between the ages of 6 months and 1.5 years.
- Late children's room. Here, the main signs of the disease can be seen only after the child is more than one and a half years old.
At the same time, the most severe form is considered to be congenital.
Werdnig-Hoffman disease. Treatment
There are no specific treatments for Werdnig-Hoffman disease. All treatment methods are aimed only at specific symptoms and manifestations.
A gastrostomy may be necessary for children who have problems feeding. Options for controlling breathing problems range from the use of non-invasive procedures to long-term invasive procedures such as a tracheotomy. Any treatment decisions should be made only after consultation of the entire medical team with the child's parents. Physical and occupational therapies are useful in minimizing contractures and developing compensatory strategies. In some cases, orthopedic devices (such as braces) and surgical correction of scoliosis may be required.
Diagnostics

Manifestations of proximal spinal amyotrophy often resemble the course of other neurological and congenital diseases, as well as traumatic injuries to the structures of the spinal cord and brain. Diagnosis of this disease is especially difficult in newborns and young children.
The key points in diagnosing spinal amyotrophy are the following studies:
- Careful history taking. The presence of cases of spinal amyotrophy in relatives allows us to suspect this hereditary disease.
- Electroneuromyography is a special study of the neuromuscular system. In this case, primary muscle damage is excluded and signs indicating pathology of the motor neurons of the spinal cord are identified.
- Computed and magnetic resonance imaging. These methods sometimes make it possible to detect atrophic changes in the anterior horns of the spinal cord. However, more often they are used to exclude other pathologies from the structures of the spinal column and brain.
- Muscle biopsy followed by histological examination of the biopsy sample. Specific muscle changes are revealed, consisting in the alternation of bundled atrophic and unchanged muscle fibers. In addition, compensatory hypertrophied muscle areas can be detected, as well as replacement of muscle tissue with connective tissue.
- Genetic analysis. Allows you to identify the exact cause of the disease: DNA testing reveals a gene mutation on the fifth chromosome.
If there have been cases of the birth of children with spinal amyotrophy in the family, when planning a subsequent pregnancy, the married couple is sent for consultation with a geneticist. Prenatal fetal DNA testing is also mandatory. Detection of Werdnig-Hoffman syndrome at the stage of prenatal diagnosis serves as an indication for termination of pregnancy.