Not to be confused with Pick's disease.
| Niemann-Pick disease | |
| ICD-10 | 75.275.2 (ILDS E75.230) |
| ICD-9 | 272.7272.7 |
| DiseasesDB | 9016, 34341 and 33390 |
| MedlinePlus | 001207 |
| MeSH | D009542 |
Niemann-Pick disease
(eng. Niemann-Pick disease) is a hereditary disease caused by a disorder of lipid metabolism and the accumulation of lipids primarily in the liver, spleen, lungs, bone marrow and brain. The disease belongs to lysosomal storage diseases and is characterized by autosomal recessive inheritance. There are three types of the disease: types A, B and.
Development of the disease
The capabilities of pathological anatomy allowed scientists to study the disease as thoroughly as possible. They were able to find out that with the development of this pathology, a metabolic disorder associated with lipids occurs. The result is the deposition of fats in the liver, spleen, lymph nodes and other internal organs. If in a healthy person they are broken down, then in a patient with Niemann-Pick syndrome they become more and more numerous, which is why the disease is classified as a storage disease.
The pathogenesis of the disease is associated with genetics. It is this that causes a person to develop such a problem. What is important is that the syndrome manifests itself already in childhood. If you notice it in a timely manner, the chances of a positive outcome will be slightly higher. The disease develops due to genetic defects of chromosomes 11 or 14 and 18, depending on the specific type of disease. With such disorders, there is a decrease in the functional activity of sphingomyelinase, which destroys sphingomyelin, which is a type of fat. For this reason, intracellular lipid transport is disrupted, the breakdown of fat deposits stops, and cholesterol levels sharply increase. Such metabolic failures are extremely dangerous to human health.
Inheritance of the disease can occur on both sides at once. If both parents had pathological genes, then the child will face a much more severe form of the disease. In such cases, a lot of effort will have to be made to save his life for at least a few years.
Both women and men can get sick. Gender has no effect on the likelihood of developing Niemann-Pick syndrome.
Niemann-Pick disease
Niemann-Pick disease
is a rare hereditary disease characterized by the accumulation of lipids in various organs and tissues, which leads to disruption of their functions. A distinctive feature is pronounced clinical polymorphism.
The most common are focal neurological symptoms, delayed neuropsychic development, hepato- and splenomegaly. Diagnostics uses determination of the activity of specific enzymes, histological studies, cerebral tomography, and molecular genetic analysis.
Symptomatic, substrate-reducing therapy is used for treatment.
Niemann-Pick disease (NPD, sphingomyelinosis, sphingomyelin lipidosis) belongs to the group of lysosomal storage diseases. The nosology was first described by the German pediatrician A. Niemann in 1914; in 1930, the German pathologist L. Pick published pathomorphological data.
There are 3 types of the disease, differing in pathogenesis, epidemiology, and nature of the course. The prevalence of types A and B in the general population is 1 case per 250,000 people, type C - 1 case per 120-150,000 people.
Among Ashkenazi Jews, type A is much more common, according to various sources, 1:40,000-1:100,000.
Niemann-Pick disease
The occurrence of all types of Niemann-Pick disease is based on genetic mutations. Types A and B are caused by a mutation in the SMPD-I gene, located at the 11p15.4-p15.1 locus. This gene encodes the enzyme acid sphingomyelinase.
Type C is caused by mutations in the NPC1 (18q11-q12 locus) and NPC2 (14q24 locus) genes. These genes encode carrier proteins involved in the transport of cholesterol and other lipids within the cell.
The pathology is inherited in an autosomal recessive manner.
The mechanisms of disease development at the pathogenetic level for different types of Niemann-Pick disease are somewhat different. As a result of a genetic mutation in BNP-A, an almost complete deficiency of acid sphingomyelinase occurs, which leads to the rapid accumulation of sphingolipids in the central nervous system and other internal organs.
This is accompanied by the rapid development of severe neurological symptoms and death in early childhood. Another type of mutation of the same gene in type B causes only a 20% decrease in the functional activity of sphingomyelinase. Therefore, lipid deposition occurs predominantly in the cells of the reticuloendothelial system (liver, spleen).
In Niemann-Pick disease type C, due to disruption of transporter proteins, different classes of lipids accumulate in cells - non-esterified cholesterol, sphingomyelin, glycosphingolipids. The nervous system and internal organs are affected. Cholesterol aggregations cause a secondary decrease in sphingomyelinase activity by inhibiting its synthesis.
In clinical practice, there are 3 main types of BNP:
- Type A (classic infantile).
The most severe form. It is characterized by an early onset, progressive course, and rapid onset of death. - Type B (visceral).
Typically more moderate course, late onset. Neurological symptoms are practically absent. - Type C.
The most common type with extremely varied symptoms. Depending on the age of onset of manifestation, it is divided into the following forms:
- neonatal – up to 3 months;
- early infantile – from 3 months to 2 years;
- late infantile – from 2 to 6 years;
- youth (juvenile) – from 6 to 15 years;
- adult – over 15 years old.
The first signs appear almost from birth - an enlargement of the liver, spleen, and lymph nodes. From 4-6 months the child’s appetite decreases, nausea and vomiting occur.
Basic skills such as holding one's head, walking, and speaking are significantly delayed. In the second year of life, muscle spasticity develops.
With deep neurodegenerative damage to the brain, breathing and heartbeat disorders develop, which is the main cause of death.
Surgery
Surgical interventions were successful only in the case of Niemann-Pick-B disease.
Bone marrow stem cell transplantation in some patients can reduce the degree of visceral symptoms - hepatosplenomegaly, interstitial lung damage.
For hypersplenism with pancytopenia, splenectomy is performed. The development of liver cirrhosis with severe liver failure is an indication for liver transplantation.
Experimental treatment
Research continues to find effective treatments for Niemann-Pick disease. In experiments on laboratory mice, under the influence of gene therapy, sphingomyelinase activity in cells increased. Currently, the drug 2-hydroxypropyl-betacyclodextrin and enzyme replacement therapy for BNP type B are at the stage of clinical trials.
Palliative treatment
At a late stage of the disease with an advanced neurodegenerative process, measures are taken to alleviate the patient’s condition. If swallowing is severely impaired, tube feeding or a gastrostomy tube may be necessary to provide the patient with sufficient nutrients and fluids.
In most cases, the prognosis for life with Niemann-Pick disease is unfavorable. Type B is considered relatively benign, in which the nervous system is not affected.
With type A, life expectancy is 1-4 years, with type C - about 10-20 years from the moment of diagnosis.
Less commonly, death occurs from severe respiratory tract infections and liver failure. The main method of primary prevention is prenatal diagnosis in the early stages of pregnancy. In the chorionic villi, molecular genetic tests determine the presence of mutations NPC1, NPC2, SMPD-1; sphingomyelinase activity is studied in amniocytes.
Source: https://www.KrasotaiMedicina.ru/diseases/genetic/Niemann-Pick
Forms and manifestations
There are several types of this disease, each of which differs in the characteristics of changes within the body, symptoms and prognosis. Most doctors tend to distinguish only 3 forms:
- Type A – classic (infantile);
- Type B – visceral (chronic);
- Type C – juvenile (subacute).
Some additionally tolerate type D, which is extremely rare. However, most often it is classified as a juvenile form, because they are almost identical.
Type A
The infantile form of the disease has an unfavorable prognosis and damages the nervous system. The first signs of its development appear already in the first year of the baby’s life. Moreover, immediately after birth the baby looks quite healthy, but after a few weeks the manifestations of the disease make themselves felt.
The baby may not hold his head up, may not roll over from his tummy to his back, may not show any attention to toys, he may have increased muscle tone in the limbs, combined with weakness, his mouth is often constantly open, which leads to drooling. Gradually, the child may completely lose hearing and vision may deteriorate. Sometimes babies develop.
After some time, parents may notice short stature, pronounced apathy, an enlarged abdomen associated with the growth of the liver and spleen, as well as ascites, the child’s arms and legs may become very thin, and the skin is dry, and sometimes it is covered with yellowish spots. It is often possible to identify defects in the cornea, retina or lens. Sudden jumps in body temperature are possible.
Almost everyone with this diagnosis does not even live to be 3 years old.
Type B
The visceral form of the disease is more favorable. Its first manifestations can be noticed when the child is between 2 and 6 years old. The main difference from the first type of Niemann-Pick syndrome is the absence of damage to the nervous system.
The disease begins with an enlarged spleen. A little later, the liver also begins to grow, which causes high bleeding, anemia, pain inside the abdominal cavity, nausea and stool disorders. In most cases, you will notice a slight increase in the abdomen. Many patients often experience colds, because... infiltrates form in their lung tissues.
Many people suffering from this type of disease live into adulthood, sometimes even into old age. However, this result will require regular maintenance of your health by taking medications, as well as an emphasis on a healthy lifestyle.
Type C
The subacute form of Niemann-Pick syndrome is also called adolescent. She is unfavorable. Parents may notice the first symptoms when their child is between 2 and 5 years old. The disease reaches its peak by age 15. Its peculiarity is a violation of sphingomyelin transport.
First, the child's muscle tone decreases, and then increases. After some time, severe weakness appears, the functions of the eyeballs malfunction, the movement of both eyes becomes uncoordinated, coordination is impaired, tremors of the limbs appear, and it becomes difficult to swallow and speak. Almost all patients gradually lose mental skills, become unable to learn, and develop dementia. The most noticeable symptoms of Niemann-Pick disease type C may be manifestations of torsion dystonia, epileptic seizures, disruption of the pelvic organs, yellowing of the skin, and retinal pigmentation.
Patients can live up to 15-18 years, after which they die. In the absence of the necessary medical care and support from loved ones, the prognosis will be even more negative, and life expectancy will be reduced by several years.
Diagnosis, treatment
To start taking medications, you must undergo diagnostic testing to confirm the diagnosis. Only after this the doctor will prescribe all the necessary medications. This will not take much time, but you should not delay visiting the hospital if symptoms appear.
Diagnostics
The child’s parents must be present at the doctor’s appointment, because It is on the basis of their words that he will be able to make a preliminary diagnosis, as well as determine the need for certain diagnostic procedures. Immediately after the survey, the child will receive all the necessary directions. If he is admitted to the hospital with serious symptoms, then all diagnostics can be carried out on the first day.
To identify Niemann-Pick syndrome, the following procedures may be necessary:
- Studying the family tree for the presence of the disease in relatives.
- A blood test to check the amount of sphingomyelinase.
- Biopsy of internal organs (liver, spleen or lymph nodes) to study fat.
- Genetic studies to analyze changes within genes.
Sometimes other procedures may be additionally required, but most often the examination is limited to those indicated above. It is very important to prepare the child in advance for them if the doctor asks to follow certain conventions.
Treatment
Any clinical case with this disease is incurable. However, you still need to take medications. This is especially true for those people who suffer from type B syndrome. Only proper treatment will help them save their lives. Its main focus is eliminating symptoms, but this does not diminish its importance.
Patients are prescribed:
- Antidepressants;
- Anticonvulsants;
- Anti-infective agents;
- To dilate the bronchi;
- Anticholinergic substances;
- Strengthening against diarrhea;
- Correcting salivation;
- Vitamin complexes.
Very often, patients are prescribed Miglustat. This drug allows you to reduce the activity of sphingomyelin production and prevent destruction of the nervous system, which will slow down the course of the disease. Doctors also advise patients to lead a healthy lifestyle, paying special attention to their health.
Sometimes an emergency blood transfusion or intravenous albumin may be required.
How to protect yourself
Niemann-Pick disease in children is a very dangerous phenomenon that almost always leads to tragic consequences. There are no significant recommendations to protect yourself. You just need to check with a geneticist when planning a pregnancy to determine the risks of developing the syndrome in your baby. There are no other ways to avoid this disease.
Niemann-Pick disease belongs to a group of genetic diseases and is quite rare. The risk of developing this genetic disease increases in closely related marriages.
There are several variants of the development of the disease, which differ in the severity of manifestations. The three most common types of disease are:
- type A (classic form),
- type B (visceral form (main manifestations of the internal organs)),
- type C (chronic adolescent form).
What happens in the body with Niemann-Pick disease?
Niemann-Pick disease is a hereditary disease and develops due to mutations in genes that are responsible for certain body functions. With this pathology, the structure of the gene that is responsible for the synthesis of the enzyme sphingomyelinase is disrupted. Pick's disease is also called sphingomyelinosis, since the basis of all disorders is a defect in the substance of the same name.
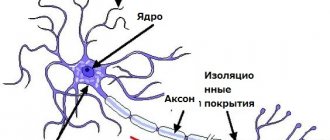
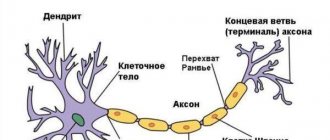
The enzyme sphingomyelinase is involved in the breakdown of lipids and other products of fat metabolism. The main task of this substance is to separate a lipid called sphingomyelin into smaller particles. Since the required enzyme is secreted in insufficient quantities, or even not synthesized at all, the lipid sphingomyelin begins to endlessly accumulate in the body. The lipid accumulates mainly in the cells of the liver, spleen, lymph nodes and in the nerve cells of the spinal cord and brain.
As these substances accumulate, the cells enlarge and stop working properly. The accumulation of lipids in nervous tissue leads to their destruction and further transformation into scar tissue, which cannot perform the necessary functions.
Pick's disease - symptoms of the disease
Clinical manifestations depend on the type of disease. Different types have their own characteristics and severity of the disease. Life expectancy also differs with different forms of gene dysfunction.
Type A
Niemann-Pick disease type A is an inherited disease that most often occurs in Jews from Central and Eastern Europe. In this variant of the disease, the lipid sphingomyelin is not broken down at all and quickly fills the cells, causing disruption of their functions. The cells increase in size and quickly die, being replaced by scar tissue.
With Pick's disease, symptoms appear in infancy - at 3-5 months. These babies have difficulty feeding, do not gain weight, and are stunted. The belly of such children is proportionally larger than the body due to the enlargement of the liver and spleen. The main symptoms of Pick's disease type A:
- early onset of the disease (3-5 months),
- vomiting, frequent diarrhea,
- temperature increase,
- weight loss, muscle atrophy, lethargy,
- The volume of the abdomen constantly increases,
- delayed psychomotor development (the child does not touch his fingers, does not sit down),
- convulsions,
- vision is impaired, children do not fix their gaze,
- hearing decreases, children react poorly to sounds.
Type A is characterized by rapid progression of disease symptoms. The affected cells die, and the death of brain cells causes problems with swallowing, breathing and circulation. Such children rarely live beyond 3-5 years.
Type B
A feature of the course of Pick's disease type B is the lack of lipid accumulation in nerve cells. Thus, nerve cells are not destroyed and symptoms of brain dysfunction do not appear. In these children, intellectual abilities do not suffer; in some cases, patients demonstrate quite high mental talent.
The first symptoms of the disease appear after 3 years. First of all, the spleen and later the liver begin to enlarge in children. With age, symptoms of lung damage appear. The lipid accumulates in the lymph nodes and causes a decrease in the activity of the immune system; children often get sick. The life expectancy of such patients is somewhat reduced, but they live to adulthood, sometimes even to old age.
Manifestations of Niemann-Pick disease type B:
- increase in abdominal volume (due to enlargement of the liver and spleen),
- periodic dull pain in the abdomen,
- nausea, sometimes vomiting,
- dysfunction of the liver and gallbladder (yellowness of the skin and eyes),
- increased bleeding (the liver does not produce components for blood clotting in sufficient quantities).
- shortness of breath during moderate physical exertion,
- frequent respiratory infections and colds.
Type C
Type C Niemann-Pick disease appears after the first years of life. At the onset of the disease, damage to internal organs occurs - enlargement of the liver and spleen, lymph nodes.
With this variant of the sphingomyelin lipid metabolism disorder, both internal organs and the nervous system are affected.
Symptoms of damage to internal organs:
- increase in abdominal volume,
- aching, dull pain in the abdomen,
- yellowness of the skin, mucous membranes and eyes,
- enlarged and painful lymph nodes,
- dyspnea,
- frequent bronchitis and pneumonia.
As the disease progresses, symptoms of damage to the nervous system appear. Manifestations of disorders of the brain and spinal cord are constantly increasing, patients lag behind their peers in mental and physical development. As the disease progresses, children lose skills and abilities that they have already mastered. For example, a child has already learned to speak, but over time, speech becomes disrupted and becomes less intelligible.
Damage to cells of the nervous system constantly progresses and causes disorders incompatible with life. Typically, such patients live 15-20 years.
Symptoms of nervous system damage:
- tremor of fingers, loss of coordination of movements,
- convulsions, epileptic seizures,
- swallowing and breathing disorders,
- loss of speech and other acquired skills,
- memory and thinking disorders, decreased school performance,
- behavioral disorders, withdrawal,
- emotional instability, irritability, depression.
Types of disease
Types A and B
Type A
- the most severe type, which begins in infants and is characterized by an enlargement of the liver and spleen (hepatosplenomegaly) and progressive damage to the nervous system. At the same time, children do not survive the early childhood period. The most common occurrence of this type of Niemann-Pick disease is observed in Ashkenazi Jews (immigrants from Central and Eastern Europe) - approximately 1 case in 40 thousand.
More moderate type B
includes hepatosplenomegaly, growth retardation, and impaired pulmonary function with frequent pulmonary infections. Other indicators include elevated cholesterol and blood lipids, and a low platelet count (thrombocytopenia). Patients usually survive to adulthood.
Types A and B are caused by mutations in the lysosomal acid sphingomyelinase gene ( SMPD1
). This enzyme is responsible for the breakdown of sphingomyelin in lysosome membranes. Its deficiency leads to excessive accumulation of sphingomyelin, and as a consequence to a broader disruption of lipid metabolism, including the accumulation of cholesterol and other cell lipids.
Type C
Type C
manifests itself in childhood, although onset may occur in infancy or in adults. Symptoms include severe liver problems, breathing problems, developmental delays, seizures, increased muscle tone (dystonia), and poor coordination, feeding, and vertical eye movement. Patients survive to adulthood. The incidence of the disease is 1 in 150 thousand.
This type of Niemann-Pick disease is caused by mutations in the NPC1
or
NPC2
, which encode a cell membrane protein responsible for the transport of cholesterol and lipids within the cell.
Diagnosis of the disease
When complaining about an increase in the volume of the child’s abdomen, or delays in mental and physical development, the doctor asks the parents in detail about the appearance of symptoms, their frequency and connection with various external factors. The doctor carefully examines the child and, if he suspects he has a genetic disease, prescribes additional tests. Also, such a child must be shown to a geneticist.
Tests to confirm Niemann-Pick disease:
Treatment
Depending on the type and clinical manifestations, the doctor selects therapy aimed at reducing the severity of the disease. The treatment regimen consists of many different drugs that improve the functions of the liver and spleen, and the outflow of bile. Drugs are also prescribed to improve the functioning of the nervous system. The use of a complex of vitamins and minerals is indicated.
Patients must be prescribed a diet with limited consumption of certain foods, such as brown bread, corn, juices, potatoes.
The following were completely banned:
- dairy products;
- White bread;
- cabbage;
- sweets;
- carbonated drinks;
- jam;
- legumes;
- cucumbers
Without restrictions, you can eat buckwheat, all types of meat, eggs, seafood, vegetables and unsweetened fruits. Sweet foods include honey, herbal teas, glucose, and fructose.
Doctors have not yet been able to completely cure the disease, but properly selected therapy can significantly reduce the severity of symptoms and improve the patient’s quality of life.
This disease is otherwise called sphingomyelinosis. It refers to hereditary pathologies and is associated with the accumulation of excess fat in the tissues of the liver, brain and bone marrow, lymphatic system, and spleen. There are several variants of the disease with different prognoses. No special methods have yet been developed to combat this disease, but medical science does not stand still. There are already positive results of therapy for Niemann-Pick disease.
This disease is of genetic origin. The likelihood of sphingomyelinosis in your child is very high if there is a history of this in the family or if your marriage is closely related. In this regard, before planning a baby, you need to visit a gynetic doctor.
The cause of the development of this pathology is disturbances in chromosomes 11, 14 and 18. Such changes affect the activity of the enzyme responsible for the breakdown of sphingomyelin, which is a type of fat, which leads to disruption of metabolic processes in the body.
Cholesterol and sphingomyelin accumulate in tissue microphages - cells of the reticuloendothelial system. Such microphages are most found in the central nervous system, lymph nodes, liver, spleen, and bone marrow.
This disease affects people of both sexes. The most severe case is when there is a coincidence of pathological genes present in both parents.
general description
Niemann-Pick disease type C (NPD) is a rare inherited disease characterized by the accumulation of cholesterol and glycosphingolipids primarily in the brain, leading to severe neurological, mental and systemic disorders.
BNP occurs in approximately one in one hundred thousand newborns. The disease is widespread. Most patients die in childhood and adolescence. LBP is caused by mutations in the NPC1 gene (approximately 95% of cases) or the NPC2 gene (approximately 5% of cases), which leads to disruption of intracellular lipid transport and abnormal storage of cholesterol and glycosphingolipids in the brain and other organs. This process leads to progressive mental disorders, damage to nerve nodes, and enlargement of the liver and spleen. BNP is a neurovisceral disease, which is characterized, first of all, by dysfunction of the central nervous system, and secondly, of internal organs. Inherited in an autosomal recessive manner.
Symptoms
The development of Niemann-Pick disease can occur in several ways. Four have been fairly well studied:
- Type A is the classic form of pathology, acute neuropathic or infantile. Symptoms appear during the first year of life. Children most often die before the age of 3.
- Type B – visceral form. The first signs appear when the child is between 2 and 6 years old. The possibility of death is lower. Some patients live to old age.
- Type C – non-acute teenage form. Symptoms of the pathology may first appear at 2-5 years of age. The intensity of the disease increases by the age of 15-18 years. Most often, children die at this age.
- Type D has been identified in Canadian residents and is completely identical to type C.
Type A
This form can be called the most unfavorable. The first symptoms of the pathology become noticeable within a few weeks after the baby is born. The baby loses appetite, weight and is stunted. Diarrhea and vomiting occur frequently. Due to the increase in the size of the liver and spleen, the child’s tummy grows. Against the background of a large belly, the arms and legs look thin and thin.
The baby's skin becomes dry and less elastic. It acquires a yellowish tint with spots of gray and brown. Upon palpation, enlarged lymph nodes are felt.
A dark red spot forms on the retina of the fundus. Sometimes the cornea becomes cloudy and the lens turns brown.
The first signs of dysfunction of the nervous system appear, which are manifested by a delay in the child’s development: he cannot hold his head, is not interested in toys, and does not roll over. There is an increase in muscle tone in the limbs, which leads to muscle weakness, and a change in tendon reflexes occurs. The child loses vision and hearing, and epilepsy attacks are possible.
At the height of the illness, the baby almost does not react to what is happening, he is lethargic and apathetic. His mouth is open all the time, from which saliva flows.
Sometimes hypothermic crises occur, that is, the temperature suddenly rises.
Children with this form of pathology become exhausted over time and die by the age of 4.
Type B
This form of the disease has a more favorable prognosis. It does not damage the nervous system. Cholesterol and sphingomyelin accumulate in the tissues of internal organs. Doctors still have not figured out why the nervous system is not affected.
By the age of 2-6 years, the baby’s spleen enlarges significantly. Later the same thing happens to the liver. Its defeat leads to a violation of blood clotting, which is expressed in the appearance of bleeding, and anemia may develop. The functioning of the gastrointestinal tract is disrupted: vomiting, nausea, and diarrhea occur. The child often has pain in the tummy, which increases in size, although not as much as with form A.
Fats accumulate in the lung tissues, and infiltrates form. This causes frequent colds.
This form of Niemann-Pick disease is characterized by a long, chronic course. Patients live to adulthood and have a much longer life expectancy.
Type C
The cause of this type of disease has not been precisely established. It is believed that it is associated with impaired transport of sphingomyelin. In this case, there is a slow accumulation of cholesterol and sphingomyelin in the liver, spleen and brain.
The first signs of the disease may appear between the ages of 2 and 20 years. The spleen and liver do not enlarge very much compared to types A and B. The skin turns yellow. A spot forms on the fundus of the eye - a “cherry pit”. The pigmentation of the retina of the eye changes.
The first manifestation of neurological disorders is a decrease in muscle tone, which increases over time. Paresis of a spastic nature appears. The muscles weaken and their tone increases.
Coordinated eye movement is impaired, especially noticeable when looking up. Vertical ophthalmoparesis develops.
Due to lack of coordination, gait changes. Uncontrolled movements of the limbs and their trembling occur. Tersion dystonia develops, expressed by violent twisting movements of the torso and head. Epilepsy seizures occur. Swallowing and speech are impaired.
There is a gradual destruction of mental abilities, which leads to the development of dementia. Control over the functioning of the pelvic organs is lost. Sometimes with this disease, strong emotions can lead to loss of muscle tone in the jaw, neck, and legs.
The disease gradually progresses and as soon as it reaches its peak, the patient dies.
Type D
Identified in residents of Canada, the province of Nova Scotia. The reason is not entirely clear, but the pathology develops with a small accumulation of sphingomyelin, while there is a lot of cholesterol.
The manifestations of the disease exactly repeat type C. Some scientists do not distinguish it into a separate form.
Pick's disease: causes, symptoms and diagnosis, treatment in children
Pick disease (Niemann-Pick disease) is a familial disease that is inherited. The pathology is manifested by excessive accumulation of adipose tissue in internal organs, including the brain. Pick's disease has several clinical forms.
Causes and characteristics of the disease
Pick's disease develops as a result of the fact that certain enzymes are not active enough, and this causes the accumulation of metabolic products, in this case fats. Normally, excess fat is broken down and eliminated from the body, which does not happen in patients with this disease.
There are four forms of this disease - type A, B, C, D. Each form of Niemann-Pick disease is caused by a mutation in a specific set of chromosomes.
The defect can be observed in the 11th, 14th or 18th chromosome.
The result of such a deviation from the norm is a violation of the breakdown of sphingomyelin, resulting in metabolic disorders and the accumulation of fats in the tissues of internal organs.
Inheritance method
The pathology is inherited in an autosomal recessive manner. Thus, if one of the parents is sick, the child will inherit the pathology, regardless of gender.
The most unfavorable case is considered to be if both parents are carriers of the pathological gene. In this case, the child's Pick's disease will be more severe. As a rule, the prognosis is unfavorable.
Types of disease
Symptoms, development and prognosis largely depend on the type of disease. There are four types of Niemann-Pick disease:
- classic form (type A);
- visceral type (type B);
- juvenile form (type C);
- a special form of Nova Scotia (type D).
The classic form is characterized by acute neuropathic development. In Niemann-Pick disease of the visceral type, a chronic course is observed, but the nervous system is not involved in the pathological process.
The juvenile form of Pick's disease type C is characterized by a subacute development with involvement of the nervous system.
Type D is found only among residents of Nova Scotia (Canada), so in many sources it is combined with the juvenile form due to the similarity of symptoms.
Symptoms of pathology
Each form of Niemann-Pick disease has its own symptoms and developmental features.
The classic form of pathology is considered the most unfavorable. Children are born absolutely healthy, but the pathology progresses rapidly during the first weeks of life. Then the following symptoms appear:
- lack of appetite;
- constant nausea to vomiting;
- stunted growth, rapid weight loss;
- increase in liver size;
- increase in spleen volume;
- skin pigmentation;
- enlarged lymph nodes;
- clouding of the cornea of the eye.
The patient's abdomen looks very large due to the increase in the size of the internal organs.
The next stage in the development of the disease is damage to the central nervous system, which is manifested by developmental disorders, lack of speech, hearing and vision impairment. The reflex activity of patients is greatly increased. It is possible to develop convulsive seizures, as with epilepsy.
This form of pathology is the most severe; death occurs between the ages of one and five years. The cause of death is usually exhaustion.
The second type of disease (type B) has a favorable course. With this form of Pick's disease, the symptoms are as follows:
- increase in the size of the spleen;
- increase in liver size;
- blood clotting disorder;
- anemia;
- indigestion.
An increase in the size of internal organs is diagnosed in the sixth year of a child’s life. An enlarged liver leads to impaired blood formation, which often results in anemia.
External manifestations are expressed in an increase in the size of the abdomen, but the pathology is not as noticeable as with the first type of disease. Patients often experience nausea and vomiting, stomach pain and constipation.
Patients with this form of pathology often catch colds.
The third type of pathology (type C) often manifests itself in adolescence to 20 years. This form is characterized by the following characteristics:
- slightly enlarged internal organs;
- visual impairment;
- skin pigmentation;
- decreased muscle tone;
- spastic paresis;
- impaired rotation of the eyeball;
- change in gait;
- tremor of fingers;
- progression of mental retardation;
- swallowing dysfunction.
Over time, the symptoms are accompanied by weakness of the leg muscles, due to which the patient is unable to walk. The prognosis for Pick's disease type C largely depends on the symptoms observed in the patient. Death occurs quickly when multiple symptoms are present. Pick's disease type D has the same symptoms, but differs in the development process.
Treatment of pathology
It is impossible to cure the pathology. Drug treatment is aimed at reducing symptoms. For this purpose, use:
- anti-seizure medications;
- antidepressants;
- drugs against diarrhea and to improve gastrointestinal function;
- drugs against muscle spasms and muscle tremors;
- antibiotics and cold medicines.
To slow the progression of symptoms of damage to the nervous system, patients are shown the drug Miglustat. Therapy with this drug allows you to stop the progression of the disease and prolong the patient’s life for many years. A positive effect is observed only with long-term use of the medicine, so the therapeutic course lasts at least six months.
https://www.youtube.com/watch?v=OU34V1-Jric
To maintain the function of the affected organs, blood transfusions are prescribed. To support the vital functions of the body, injections of vitamin preparations are recommended.
Forecast
Pick-Niemann syndrome is an incurable disease that is fatal. The most severe form of pathology is the first type, in this case death occurs during the first years of life.
In other cases, patients can live up to 30 years. The prognosis depends on the treatment and the therapeutic methods used. The development of the disease occurs rapidly. The peak development of pathology occurs during the period of damage to the nervous system. At this time, there is a rapid regression of the patient’s mental development, hearing loss, and deterioration of vision.
There are no preventive measures for Niemann-Pick disease. Families whose relatives have this syndrome need to be examined by a geneticist before planning a pregnancy.
Source: https://NashiNervy.ru/tsentralnaya-nervnaya-sistema/simptomy-bolezni-pika-u-detej-i-ee-osobennosti.html
Treatment
Depending on the form of the disease and the severity of its course, the doctor will prescribe treatment to reduce the baby’s suffering. Therapy includes drugs that improve the functioning of the spleen and liver, and the outflow of bile. In addition, medications are used that have a positive effect on the functioning of the nervous system. Complexes of minerals and vitamins are prescribed.
In addition to the medications listed, a diet that limits corn, juices, potatoes, and brown bread is usually prescribed.
The following are completely prohibited:
- White bread;
- dairy products;
- cabbage;
- carbonated drinks;
- sweets;
- cucumbers;
- jam;
- legumes.
Not included in the prohibited list: meat, eggs, buckwheat, seafood, unsweetened fruits and vegetables not listed in the first list. You can use fructose, glucose, honey.
But doctors do not have a unified opinion on diet. Some of them consider proper nutrition to be the basis for treating the disease. Others argue that diet is powerless for genetic disorders. Following a diet will bring additional suffering to a sick child.
Important! Niemann-Pick disease has no cure. The right treatment simply reduces the severity of symptoms and improves the patient's life.
Niemann-Pick disease (sphingomyelinosis) is a hereditary disease associated with excessive accumulation of fats in various organs and tissues, primarily in the brain, liver, lymph nodes, spleen, and bone marrow. Has several clinical variants, each with a different prognosis. There is currently no specific treatment. From this article you can learn about the cause, symptoms and treatment options for Niemann-Pick disease.
Niemann-Pick disease is a lysosomal storage disease. This is when, as a result of insufficient activity of an enzyme, intermediate metabolic products accumulate in the cells of the body, which normally undergo further breakdown.
Causes of Niemann-Pick disease
The disease is based on a genetic defect of the 11th chromosome (types A and B), 14th and 18th chromosomes (type C). As a result of the presence of a disorder in the gene structure, a person experiences a decrease in the activity of the enzyme sphingomyelinase, which breaks down sphingomyelin. Sphingomyelin is a type of fat. This biochemical disorder leads to excessive accumulation of sphingomyelin and cholesterol in the cells of the reticuloendothelial system: tissue macrophages. As a result, metabolism is disrupted.
Tissue macrophages are scattered throughout the body, but most of them are in the spleen, liver, bone marrow, lymph nodes, and central nervous system.
The disease is autosomal recessive in nature, that is, it is not associated with gender; both men and women can be affected. When two pathological genes coincide (from the father and from the mother), the disease is most severe.
Niemann–Pick disease – clinical picture
This disease is otherwise called sphingomyelinosis. It refers to hereditary pathologies and is associated with the accumulation of excess fat in the tissues of the liver, brain and bone marrow, lymphatic system, and spleen.
There are several variants of the disease with different prognoses. No special methods have yet been developed to combat this disease, but medical science does not stand still.
There are already positive results of therapy for Niemann-Pick disease.
This disease is of genetic origin. The likelihood of sphingomyelinosis in your child is very high if there is a history of this in the family or if your marriage is closely related. In this regard, before planning a baby, you need to visit a gynetic doctor.
Causes of Niemann-Pick disease
Niemann-Pick disease is genetic in origin
The cause of the development of this pathology is disturbances in chromosomes 11, 14 and 18. Such changes affect the activity of the enzyme responsible for the breakdown of sphingomyelin, which is a type of fat, which leads to disruption of metabolic processes in the body.
Cholesterol and sphingomyelin accumulate in tissue microphages - cells of the reticuloendothelial system. Such microphages are most found in the central nervous system, lymph nodes, liver, spleen, and bone marrow.
This disease affects people of both sexes. The most severe case is when there is a coincidence of pathological genes present in both parents.
Complications
Possible complications with this disease: deafness, blindness, mental retardation, late development of motor abilities.
Symptoms
There are several clinical variants of Niemann-Pick disease. The division into variants is due to the characteristics of the course and biochemical changes.
A total of 4 types of disease have been studied:
- type A – classic form of the disease (infantile, acute neuropathic);
- type B – visceral form (chronic, without involvement of the nervous system);
- type C – juvenile form (subacute, chronic neuropathic);
- type D is a form from Nova Scotia (named after the province in Canada in which this form is found). Recently, this type has been combined with type C.
Type A
This is the most unfavorable form in terms of prognosis for life. It manifests itself a few weeks after birth (at birth, children look healthy). The child's appetite worsens, he begins to lose weight and lag in growth. Periodic vomiting and diarrhea are possible. The abdomen gradually enlarges due to the liver and spleen (the liver enlarges earlier than the spleen), and ascites develops. The limbs look thin and very thin compared to the enlarged abdomen.
The child's skin becomes dry, loses its elasticity, acquires a yellowish color, and yellowish-gray or yellow-brown spots are visible in places. All groups of lymph nodes are enlarged, which can be determined by palpation.
When examining the fundus, a specific symptom of “cherry pit” is determined - a dark red spot on the retina. There may be clouding of the cornea and the appearance of a brown color to the lens.
The damage to the nervous system initially consists of a lag in neuropsychic development from their peers: children do not hold their heads up, do not roll over from their stomach to their back, and do not follow the toy. Muscle tone in the arms and legs increases, and muscle weakness develops. Tendon reflexes also increase. Hearing is gradually lost, vision decreases, and epileptic seizures may occur. At the height of the disease, the child is lethargic and apathetic, weakly reacts to events happening around him, almost constantly remains with his mouth open, which is why drooling develops.
Periods of sudden increases in temperature occur: hyperthermic crises.
Exhaustion gradually develops, and patients with this form of the disease die at the age of 2-4 years.
Type B
This form of the disease has a favorable course. In this case, the nervous system is not affected; the accumulation of sphingomyelin and cholesterol occurs only in the internal organs. Why the nervous system remains intact is still a mystery to doctors.
First, the spleen enlarges, usually by 2-6 years. Later the liver enlarges. Liver damage leads to increased bleeding due to disruption of the blood coagulation system. Anemia often develops. I am concerned about abdominal pain, periodic bowel movements, and occasional nausea and vomiting. The abdomen increases in size, but not as significantly as with type A.
Due to the accumulation of fats in the lung tissue, infiltrates are formed. This causes frequent colds in such children.
This form is characterized by a long-term chronic course. Life expectancy is much longer than with type A, patients live to adulthood.
Type C
The biochemical defect in this form is not clearly understood. Sphingomyelin transport is suspected to be impaired. There is a mild accumulation of sphingomyelin and significant accumulation of cholesterol in the brain, spleen and liver.
The disease first manifests itself between the ages of 2 and 20 years. The enlargement of the liver and spleen compared to types A and B is insignificant. Characterized by a jaundiced skin tone. In the fundus there is a symptom of “cherry pit”, pigmentary degeneration of the retina.
Neurological disorders begin with a decrease in muscle tone, which then, on the contrary, increases. Spastic paresis gradually develops: muscle weakness with a simultaneous increase in muscle tone. The joint activity of the eyeballs is disrupted, coordinated eye movements become impossible, especially when looking up (the so-called vertical ophthalmoparesis).
Loss of coordination develops, causing the gait to change. Trembling and involuntary movements in the limbs occur. Characterized by violent twisting movements in the head and torso (torsion dystonia). Epileptic seizures appear. Swallowing and speech are impaired. Mental impairment gradually progresses, children lose the ability to learn, and eventually dementia develops. Control over the function of the pelvic organs is impaired. A fairly specific symptom for this form of Niemann-Pick disease has been described: a sudden loss of muscle tone in the legs, jaw and neck when laughing or other strong emotions. The disease gradually progresses.
After the appearance of a detailed clinical picture of the disease, the days of such patients are numbered.
Type D
Described among residents of the Canadian province: Nova Scotia (Scotia). A clear biochemical defect has not been identified, but the disease develops as a result of a small accumulation of sphingomyelin and a significant accumulation of cholesterol. In terms of its clinical manifestations, it is practically no different from type C, so some researchers prefer not to distinguish it into a separate form.
Niemann-Pick disease: what it is, symptoms and treatment
Niemann-Pick disease (sphingomyelinosis) is a hereditary disease associated with excessive accumulation of fats in various organs and tissues, primarily in the brain, liver, lymph nodes, spleen, and bone marrow.
Has several clinical variants, each with a different prognosis. There is currently no specific treatment. From this article you can learn about the cause, symptoms and treatment options for Niemann-Pick disease. Niemann-Pick disease is a lysosomal storage disease. This is when, as a result of insufficient activity of an enzyme, intermediate metabolic products accumulate in the cells of the body, which normally undergo further breakdown.
Causes of Niemann-Pick disease
The disease is based on a genetic defect of the 11th chromosome (types A and B), the 14th and 18th chromosomes (type C).
As a result of the presence of a disorder in the gene structure, a person experiences a decrease in the activity of the enzyme sphingomyelinase, which breaks down sphingomyelin. Sphingomyelin is a type of fat.
This biochemical disorder leads to excessive accumulation of sphingomyelin and cholesterol in the cells of the reticuloendothelial system: tissue macrophages. As a result, metabolism is disrupted.
The disease is autosomal recessive in nature, that is, it is not associated with gender; both men and women can be affected. When two pathological genes coincide (from the father and from the mother), the disease is most severe.
Symptoms
There are several clinical variants of Niemann-Pick disease. The division into variants is due to the characteristics of the course and biochemical changes.
A total of 4 types of disease have been studied:
- type A – classic form of the disease (infantile, acute neuropathic);
- type B – visceral form (chronic, without involvement of the nervous system);
- type C – juvenile form (subacute, chronic neuropathic);
- type D is a form from Nova Scotia (named after the province in Canada in which this form is found). Recently, this type has been combined with type C.
Type A
This is the most unfavorable form in terms of prognosis for life. It manifests itself a few weeks after birth (at birth, children look healthy). The child's appetite worsens, he begins to lose weight and lag in growth.
Periodic vomiting and diarrhea are possible. The abdomen gradually enlarges due to the liver and spleen (the liver enlarges earlier than the spleen), and ascites develops. The limbs look thin and very thin compared to the enlarged abdomen.
The child's skin becomes dry, loses its elasticity, acquires a yellowish color, and yellowish-gray or yellow-brown spots are visible in places. All groups of lymph nodes are enlarged, which can be determined by palpation.
When examining the fundus, a specific symptom of “cherry pit” is determined - a dark red spot on the retina. There may be clouding of the cornea and the appearance of a brown color to the lens.
The damage to the nervous system initially consists of a lag in neuropsychic development from their peers: children do not hold their heads up, do not roll over from their stomach to their back, and do not follow the toy. Muscle tone in the arms and legs increases, and muscle weakness develops.
Tendon reflexes also increase. Hearing is gradually lost, vision decreases, and epileptic seizures may occur.
At the height of the disease, the child is lethargic and apathetic, weakly reacts to events happening around him, almost constantly remains with his mouth open, which is why drooling develops.
Periods of sudden increases in temperature occur: hyperthermic crises.
Exhaustion gradually develops, and patients with this form of the disease die at the age of 2-4 years.
Type B
This form of the disease has a favorable course. In this case, the nervous system is not affected; the accumulation of sphingomyelin and cholesterol occurs only in the internal organs. Why the nervous system remains intact is still a mystery to doctors.
First, the spleen enlarges, usually by 2-6 years. Later the liver enlarges. Liver damage leads to increased bleeding due to disruption of the blood coagulation system. Anemia often develops. I am concerned about abdominal pain, periodic bowel movements, and occasional nausea and vomiting. The abdomen increases in size, but not as significantly as with type A.
This form is characterized by a long-term chronic course. Life expectancy is much longer than with type A, patients live to adulthood.
Type C
The biochemical defect in this form is not clearly understood. Sphingomyelin transport is suspected to be impaired. There is a mild accumulation of sphingomyelin and significant accumulation of cholesterol in the brain, spleen and liver.
The disease first manifests itself between the ages of 2 and 20 years. The enlargement of the liver and spleen compared to types A and B is insignificant. Characterized by a jaundiced skin tone. In the fundus there is a symptom of “cherry pit”, pigmentary degeneration of the retina.
Neurological disorders begin with a decrease in muscle tone, which then, on the contrary, increases. Spastic paresis gradually develops: muscle weakness with a simultaneous increase in muscle tone. The joint activity of the eyeballs is disrupted, coordinated eye movements become impossible, especially when looking up (the so-called vertical ophthalmoparesis).
Loss of coordination develops, causing the gait to change. Trembling and involuntary movements in the limbs occur. Characterized by violent twisting movements in the head and torso (torsion dystonia). Epileptic seizures appear. Swallowing and speech are impaired.
Mental impairment gradually progresses, children lose the ability to learn, and eventually dementia develops. Control over the function of the pelvic organs is impaired.
A fairly specific symptom for this form of Niemann-Pick disease has been described: a sudden loss of muscle tone in the legs, jaw and neck when laughing or other strong emotions. The disease gradually progresses.
After the appearance of a detailed clinical picture of the disease, the days of such patients are numbered.
Type D
Described among residents of the Canadian province: Nova Scotia (Scotia). A clear biochemical defect has not been identified, but the disease develops as a result of a small accumulation of sphingomyelin and a significant accumulation of cholesterol. In terms of its clinical manifestations, it is practically no different from type C, so some researchers prefer not to distinguish it into a separate form.
Diagnostics
To confirm the diagnosis, determine the activity of sphingomyelinase in the culture of skin fibroblasts and leukocytes (for types A and B), detect the accumulation of non-esterified cholesterol in the culture of skin fibroblasts (for type C), and search for genetic defects in the 11th, 14th, 18th chromosomes.
Bone marrow puncture in such patients reveals specific “foamy” Niemann-Pick cells (they look like this due to the accumulation of fat).
Treatment
The disease is incurable. Basically, symptomatic treatment is carried out to alleviate the suffering of the patient.
Among the symptomatic remedies used:
- anticonvulsants (Depakine and other Valproates);
- drugs to correct salivation (drip Atropine into the mouth, inject botulinum toxin into the salivary glands, use hyoscine patches);
- for mental disorders - antidepressants (selective serotonin reuptake inhibitors - Prozac, Sirlift, Zoloft) for depression and Valproate for psychosis;
- antidiarrheals: Loperamide (Imodium), diet therapy;
- with the development of infectious complications from the respiratory tract, antibiotics, bronchodilators (Berodual), and physiotherapeutic procedures are used;
- for dystonia and tremors: anticholinergic drugs (Cyclodol, Parkopan, Biperiden, Akineton).
In recent years, Miglustat has been used to stop the accumulation of sphingomyelin in cells. It blocks the enzyme responsible for the synthesis of glycosphingolipids (sphingomyelin precursors).
Used in dosages ranging from 100 mg 1-2 times a day to 200 mg 3 times a day (depending on the age and body area of the patient). Miglustat prevents the destruction of nerve cells and thus slows down the development of neurological symptoms and leads to an increase in life expectancy.
A visible positive result from using the drug develops after 6 months to 1 year of continuous use.
Thus, Niemann-Pick disease is a rather severe hereditary disease of the accumulation of sphingomyelins in the cells of the body, which leads to damage to the liver, brain, lymph nodes, and lungs.
The disease is steadily progressive. In some variants of the disease, patients quickly die, while other types have a more benign course.
A clear and effective treatment has not yet been developed, but successful steps in this direction have already been taken.
Source: https://doctor-neurologist.ru/bolezn-nimana-pika-chto-eto-takoe-simptomy-i-lechenie
Treatment
The disease is incurable. Basically, symptomatic treatment is carried out to alleviate the suffering of the patient.
Among the symptomatic remedies used:
In recent years, Miglustat has been used to stop the accumulation of sphingomyelin in cells. It blocks the enzyme responsible for the synthesis of glycosphingolipids (sphingomyelin precursors). Used in dosages ranging from 100 mg 1-2 times a day to 200 mg 3 times a day (depending on the age and body area of the patient). Miglustat prevents the destruction of nerve cells and thus slows down the development of neurological symptoms and leads to an increase in life expectancy. A visible positive result from using the drug develops after 6 months to 1 year of continuous use.
Thus, Niemann-Pick disease is a rather severe hereditary disease of the accumulation of sphingomyelins in the cells of the body, which leads to damage to the liver, brain, lymph nodes, and lungs. The disease is steadily progressive. In some variants of the disease, patients quickly die, while other types have a more benign course. A clear and effective treatment has not yet been developed, but successful steps in this direction have already been taken.
Treatment of Niemann-Pick disease
The goal of the therapy is to correct neurological and mental disorders to improve the quality of life of patients with BNP. For this purpose, symptomatic and pathogenetic agents are prescribed. Anticonvulsants, atypical neuroleptics, mood stabilizers, antibacterial drugs, and cognitive enhancement drugs are widely used. Electroconvulsive therapy has been successfully used for catatonia. Pathogenetic therapy is carried out with the drug miglustat (Zaveska), which acts as a competitive inhibitor of the enzyme glucosirceramide synthase, which catalyzes the first fixed stage of glycosphingolipid synthesis. The recommended dose of miglustat for adults and adolescents is 200 mg three times daily. The dose in patients aged 4-12 years is determined based on body surface area. At the same time, a strict diet with restriction of disaccharides is prescribed. A common complication of miglustat use is diarrhea, which requires the use of antidiarrheal drugs and probiotics. The effect of using the drug occurs no earlier than six months later, and often much later, but the prognosis is significantly improved.
Essential drugs
There are contraindications. Specialist consultation is required.
- Miglustat (Zavesca) is a glucosylceramide synthase inhibitor. Dosage regimen: taken orally, regardless of food intake, at a dose of 200 mg 3 times a day. for adults and children over 12 years old. The dose for young children is determined based on body surface area. If diarrhea or renal failure develops, the dose of the drug is reduced.