Dystrophic myotonia (Steinert's disease) is a pathology of genetic origin with slow progression, based on the presence of a defect in myotonin-protein kinase. This leads to the formation of myotonia in combination with myofiber dystrophy.
Steinert's disease is transmitted to offspring in an autosomal dominant manner. The classic version is formed mainly between the ages of ten and twenty years. And how rarity can develop in the first days of life.
Morphologically, there is a combination of hypertrophic transformations of some myofibers with atrophic ones in others, the transformation of some into fatty and connective tissue structures. Histologically, destruction of muscle fibrils and transformation of mitochondrial size occur.
1.General information
Leiden-Thomsen disease, or myotonia congenita, is a group of inherited diseases and is rare: the prevalence in the general population is estimated at one case per 200,000 population.
Congenital myotonia is inherited in an autosomal dominant manner, that is, to develop the clinical form it is enough to receive a defective gene from one of the parents. The diagnosis entered the medical nosological paradigm from 1874-1876, when familial cases of the disease were first described.
Among the patients, males predominate.
A must read! Help with treatment and hospitalization!
Diagnostic criteria
A doctor may suspect Rossolimo-Steinert-Kurshman disease if the patient has a combination of myotonic and dystrophic changes in the muscles against the background of loss of intelligence and the presence of cardiovascular and endocrine pathology.
Polysystemicity almost always indicates the genetic nature of the disease. Such patients are subject to DNA analysis and genealogical analysis to confirm the autosomal dominant inheritance of the pathology. Electrocardiography, electroneuromyography, and hormone tests are used as informative research methods.
Due to the versatility of clinical manifestations, specialists from various branches of medicine are usually involved in the diagnosis process - genetics, cardiology, endocrinology, gynecology, andrology, neurology.
Differential diagnosis is made between dystrophic myotonia and other types of similar diseases. Unlike the others, Rossolimo's disease is characterized by muscle atrophy. Often, to confirm the diagnosis, it is necessary to resort to a biopsy to determine the level of muscle protein, which is increased in the tissues of this pathology.
Antenatal diagnosis is also carried out by examining amniotic fluid.
3. Symptoms and diagnosis
As a rule, Leiden-Thomsen disease manifests itself and can be identified diagnostically in prepubertal or early puberty. A specific symptom is residual tension (spastic tone) of any muscle groups that were tense voluntarily and adequately to the situation, after which they should relax - and do not relax. Most often, such tonic muscle spasms are observed in the fingers, hands, legs, orbicularis oculi muscles, and masticatory (masticatory) muscles. Accordingly, the patient for a long time, for tens of seconds, cannot, for example, open his eyes or mouth, turn his head, move his limbs, open his palm, change his facial expression, etc.
A characteristic feature is the often observed contradiction between athletic hypertrophy, rigidity, relief of spasmodic muscle groups and actually reduced muscle strength.
Reflexological examination using a neurological hammer easily reveals tonic-spastic reactions of the fingers, tongue, etc. Of the instrumental diagnostic methods, the most informative is electromyography. A consultation with a medical geneticist is required, and a detailed family history is collected and compared with the clinical picture.
About our clinic Chistye Prudy metro station Medintercom page!
Symptomatic picture
Classic version
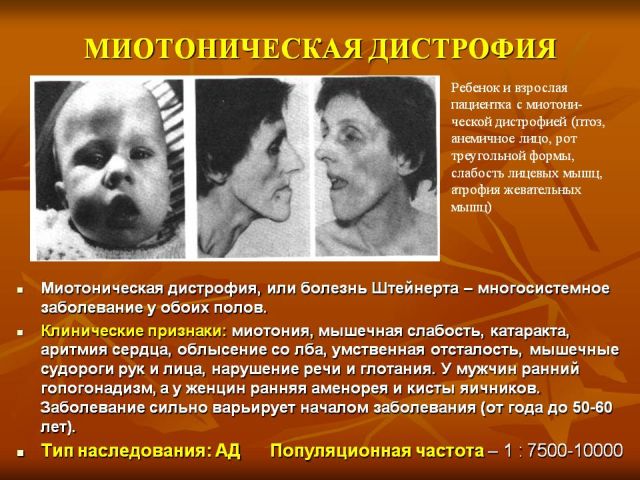
It appears after the age of five and up to more than 30 years of age. The most common age is from ten to twenty years. The picture consists of typical signs of a myotonic state with myopathy, damage to the cardiovascular system and central nervous system, a dysendocrine state and cataracts.
Myotonic symptoms include spasms, especially in the masticatory myogroups and muscles responsible for flexing the wrist. Mechanical responses are also noted, which are determined during tapping with a neurological hammer. A characteristic difference is atrophic transformations in different muscle groups. In this condition, the course of the pathological process is characterized by a slow decrease in the signs of myotonia against the background of progression of myodystrophy.
Myofibers of distant parts of the extremities, facial, sternocleidomastoid and temporal are often damaged. The involvement of facial muscles in the pathological process is characterized by the appearance of a mask-like sad face of the sick. Atrophic transformation of the pharyngeal and laryngeal myogroup leads to the formation of myopathic paresis of the larynx with vocal dysfunction and difficulties in swallowing. Pathological transformations sometimes form in the muscles of the respiratory system. In combination with spasms, they lead to pulmonary ventilation disorders, the development of sleep apnea, and pneumonia of congestive aspiration origin.
Dysfunction of the vascular-cardiac system is observed in about 50% of cases:
- arrhythmia, which is associated with conduction disorders;
- hypertrophic phenomena of the left ventricle;
- bundle branch block.
Among the manifestations of damage to the central nervous system, hypersomnia and decreased intelligence (up to a mild degree of imbecility) are often recorded.
Endocrine disorders are represented mainly by the reproductive system. In males it appears as decreased libido, cryptorchidism, impotence, etc., in females there is hirsutism, irregular menstruation in the form of oligo- or dysmenorrhea and early menopause. Characteristic is the transformation of hair structure in combination with alopecia. Men suffer from hair loss in the temporal and frontal areas, women – generalized or focal baldness.
Congenital form
Initial signs sometimes appear during intrauterine development in the form of a decrease in fetal motor activity, which is determined by ultrasound examination data in the third trimester of pregnancy.
After birth, signs of myopathy appear in the form of diffuse myohypotonia, mainly in the facial, masticatory and oculomotor muscles and in distant areas of the extremities.
This condition is characterized by difficulties with feeding and dyspnea. Myotonia appears in later periods. Characterized by mental retardation and delayed development of motor function, rapid progression of clinical signs, which often ends in death at an early age.
4.Treatment
Like most other rare diseases, Leiden-Thomsen disease is not well understood. Genetic engineering and genetic therapy methods are under development and are not yet used in clinical practice. Accordingly, therapy for diseases of this group is always palliative and is selected partly empirically, partly based on ideas about pathogenesis. To soften and reduce myotonic spasms, diacarb, diphenine, and calcium compounds (chloride or gluconate) are prescribed according to a specific regimen. Physiotherapy and physical therapy bring quite good results.
Opinions about the “natural” dynamics of congenital Leiden-Thomsen myotonia differ in different sources: one can find reports of both a steady slow progression throughout life and a tendency towards a spontaneous decrease in the severity of spastic-myotonic syndrome.
In any case, Leiden-Thomsen disease does not pose any threat to life, although, of course, it reduces the quality of life to one degree or another, causing psychological discomfort and obvious difficulties in social communication (which is a particularly painful problem at the manifest stage, i.e. in adolescence). Therefore, it is important to seek help in a timely manner, which will allow the disease to be diagnosed and adequate treatment prescribed.
Reasons for the development of Rossolimo-Steinert-Kurshman myotonia
Rossolimo-Steinert-Kurshman myotonia is always a genetically transmitted defect. The key reason for its development is considered to be a violation in the DMPK gene, located on the nineteenth chromosome. The severity of the disease is directly proportional to the number of trinucleotide CTGs. Normally, this indicator is approximately 5-37, at 50-80 a mild form of the disease is observed, at 100-500 a late severe form is observed.
Under the influence of a violation of the DMPK gene in the body, a change occurs in myotonin protein kinase - a protein localized in skeletal and muscle tissue, in the myocardium, central nervous system, etc., which provokes the appearance of myotonic spasms, combined with atrophy of the muscles of the face, neck and limbs in the distal zones. In essence, hypertrophy of some muscle fibers occurs with simultaneous atrophy of others. As a result, some of these fibers are replaced either by other fibers or by fatty or connective tissues.
Possible complications
The long course of the disease leads to the involvement of many organs and systems in the process.
Their defeat occurs indirectly - due to prolonged muscle spasm:
- Spinal curvature: forward (lordosis), backward (kyphosis), sideways (scoliosis);
- Intervertebral hernia;
- Sleep apnea syndrome (suffocation during sleep);
- Cardiomyopathy (heart damage develops because the myocardium is represented by striated muscles);
- Muscular atrophy;
- Persistent changes in facial expressions, chewing and swallowing;
- Bruxism.
REFERENCE. Course therapy allows one to avoid complications and achieve long-term remission, but patients often develop addiction to the anticonvulsant. In this regard, the possibility of self-medication and long-term use of the same drug without medical supervision should be excluded.
Symptoms of congenital Rossolimo-Steinert-Kurshman myotonia
The congenital type of myotonia can be noticed by a specialist even during the period of stay in the mother’s womb. This fetus does not show increased motor activity. Therefore, if a pregnant woman notices such behavior of the baby in the stomach, she should definitely undergo an ultrasound scan in the last three months before giving birth.
Signs of myotonia are also present in newborns. This is hypotonia of facial, masticatory muscles, distal areas of the limbs, and eyeballs. In addition, breathing disorders are also common. A little later, symptoms such as mental retardation and delayed motor development appear. The progression of these pathologies can occur at different rates. With the rapid pace of their development, the patient’s death from the disease can befall him at a very early age.
Differential diagnosis of Rossolimo-Steinert-Kurshman myotonia
It is necessary to differentiate Rossolimo-Steinert-Kurshman myotonia from other known types of myotonia, which in some situations may have similar signs and course. Atrophy of muscle tissue is characteristic exclusively of this disease and allows you to quickly distinguish it from Thomsen's myotonia, a sign of which is muscle hypertrophy. Early involvement of the facial muscles and dominant inheritance distinguish it from Becker myotonia. In addition to these conditions, it is necessary to differentiate Rossolimo-Steinert-Kurshman disease from ALS and Charcot-Marie-Tooth amyotrophy.
Etiology and pathogenesis
The development of the disease is associated with a genetic mutation that develops during the prenatal period. The damaged gene responsible for the operation of chloride channels and the synthesis of the dystrophin protein is located on the long arm of chromosome 7 . As a result of the mutation, insufficient formation or complete absence of the protein that regulates muscle contraction occurs.
Normally, dystrophin is responsible for maintaining muscle function and the correct sequence of relaxation and contraction. Due to disruption of its synthesis, chlorine is retained on the membranes of muscle cells. The striated muscle bundles lose their ability to relax and remain contracted after performing the movement.
Overvoltage of the fibers causes an even greater accumulation of chloride ions on the surface of cell membranes. The balance between the remaining ion channels is disrupted. Accumulated chlorine provokes the release of calcium ions outside the cells. Excess calcium has a direct damaging effect and leads to the destruction of muscle cells.
REFERENCE. Tonic spasms gradually cause deformation of the entire axial skeleton. Due to damage to the respiratory muscles, episodes of shortness of breath or suffocation develop. Damage to the striated muscle of the heart leads to cardiomyopathies.
Treatment of Rossolimo-Steinert-Kurshman myotonia
In modern medical practice, there is currently no therapy designed to completely cure people of Rossolimo-Steinert-Curschmann myotonia. Patients diagnosed with this disease are prescribed a diet based on foods with minimal or no potassium levels. People suffering from this myotonia should not be overcooled, since low temperatures can provoke spasms at any time.
To reduce the risk of their sudden appearance and improve the general condition of the patient, he is prescribed the following drugs: quinine, novocainamide, diphenine simultaneously with diacarb. To these medications, experts recommend adding the mandatory use of anabolic steroids: nerobol, retabolil, methylandrostenediol. It is also necessary to include small doses of ATP and B vitamins in the list of required substances.
The above drugs have a good effect in both the classical and congenital types of the disease. All of these measures can reduce the negative effects of the disease on the patient’s body and prolong his life, but they are in no way able to completely rid him of myotonia and make him an absolutely healthy person again.