Болезнь Верднига-Хоффмана – спинальная мышечная атрофия, характеризующаяся дегенерацией нервных клеток в пределах нижней области головного мозга (ствол головного мозга) и некоторых двигательных нейронов в спинном мозге (передние рога).
Более 80% младенцев испытывают тяжелую слабость уже в возрасте до 6 месяцев и практически все они никогда не достигнут способности сидеть самостоятельно. Мышечная слабость, отсутствие моторного развития и плохой мышечный тонус являются основными клиническими проявлениями этой болезни. Большинство младенцев также имеют проблемы с сосанием или глотанием. Некоторые показывают признаки брюшного дыхания в течение первых нескольких месяцев жизни. Большинство детей умирают в возрасте до 2-х лет, но выживание может зависеть от степени дыхательной функции и респираторной поддержки.
Болезнь Верднига-Хоффмана подразделяется на пять подтипов. Такое разделение основано на возрасте, в котором появляются первые симптомы / проявления, течении и прогрессировании заболевания. Подтип 0 характеризуется чрезвычайной слабостью при рождении, всем детям практически с рождения необходима немедленная искусственная вентиляция. Эти дети никогда не смогут дышать самостоятельно. Подтип 1 характеризуется острой спинальной мышечной атрофией, симптомы и признаки которой появляются в возрасте около 6 месяцев. У пациентов с подтипом 2 симптомы и проявления появляются в возрасте до 1 года. Дети с этим подтипом могут научиться сидеть, но они никогда не смогут ходить. Симптомы и проявления подтипа 3 (болезнь Кугельберга-Веландера) появляются в возрасте после 1 года. Этот подтип прогрессирует в течение определенного периода времени до потери двигательных способностей. У лиц с четвертым подтипом первые симптомы и проявления появляются в возрасте около 10 лет.
Болезнь Верднига-Хоффмана. Симптомы и проявления
Клинические особенности и скорость прогрессирования болезни Верднига-Гоффмана варьируются среди пострадавших лиц. Ранние признаки заболевания включают: мышечная слабость (часто появляется в возрасте до 6 месяцев), сниженный мышечный тонус (гипотония), ненормальная гибкость (гипермобильность) суставов, отсутствие сухожильных рефлексов и подергивание (подрагивание) языка. Чаще всего дети не обретают контроль над головой, они не могут переворачиваться, сидеть или стоять. У детей с этим заболеванием также могут возникнуть трудности с сосанием, глотанием и дыханием, они имеют повышенную восприимчивость к респираторным инфекциям и часто развивают другие осложнения, которые могут привести к потенциально опасным для жизни нарушениям в течение первых нескольких месяцев или лет жизни.
Для детей, которые, как представляется, имеют нормальное развитие в течение нескольких месяцев до появления первых признаков мышечной слабости, это расстройство, как правило, прогрессирует более медленно. По мере прогрессирования заболевания снижение мышечного тонуса и слабость могут начать постепенно распространяться почти на все мышцы, за исключением мышц, контролирующих движения глаз.
Скорость прогрессирования заболевания Верднига-Гоффмана меняется. В течение нескольких месяцев могут возникнуть проблемы с дыханием и кишечником (запоры). Ребенок может полностью потерять способность глотать. Дыхательная недостаточность или аспирация пищи также могут произойти у некоторых детей. Большинство детей умирают в возрасте до 2-х лет, но выживание может зависеть от степени дыхательной функции.
Амиотрофия Верднига-Гоффмана
Амиотрофия Верднига-Гоффмана
— это наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1-1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии.
Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани.
Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Заболевание имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю.
Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию — амиотрофию Кугельберга-Веландера.
Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Амиотрофия Верднига-Гоффмана
Амиотрофия Верднига-Гоффмана — наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы.
Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) — ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена.
Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
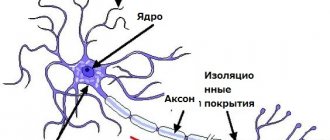
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах.
Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты.
Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
СМА I клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов.
Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают.
Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов. У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия, косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры.
Последний обуславливает развитие и прогрессирование дыхательной недостаточности, выступающей основной причиной летального исхода.
В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии, которая может явиться смертельно опасным осложнением спинальной амиотрофии.
Ранняя детская форма
СМА II дебютирует после 6-месячного возраста.
К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять.
Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов.
Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана.
Пациенты могут доживать до 15-летнего возраста.
Амиотрофия Кугельберга-Веландера
СМА III – наиболее доброкачественная спинальная амиотрофия детского возраста. Манифестирует после 2-х лет, в отдельных случаях в период от 15 до 30 лет. Отсутствует задержка психического развития, длительное время пациенты способны самостоятельно двигаться. Некоторые из них доживают до глубокой старости, не теряя способности к самообслуживанию.
Больные спинальной мышечной атрофией I типа находятся под наблюдением детских неврологов и неонатологов. Большое значение имеет возраст манифестации заболевания – для амиотрофии Верднига-Гоффмана характерно развитие с момента рождения до 6 месяцев. В анамнезе часто имеются сведения о позднем и вялом шевелении плода во время беременности.
При осмотре ребенка обращается внимание на выраженную мышечную гипотонию, неспособность самостоятельно сидеть или удерживать голову, типичную «позу лягушки» – плечи приподняты, руки вдоль туловища, ноги выпрямлены и повернуты кнаружи.
Отмечаются мышечные подергивания, ослабление или отсутствие сухожильных рефлексов, грубые костные деформации (колоколообразная грудная клетка, Х-образные нижние конечности).
Для подтверждения диагноза назначаются следующие дополнительные исследования:
- Лабораторные тесты.
В биохимическом анализе крови обнаруживается небольшое повышение концентрации креатинфосфокиназы. У некоторых пациентов данный показатель может находиться в пределах нормальных значений. При анализе газов крови выявляется снижение парциального давления кислорода (paO2) и увеличение углекислого газа (paCO2). - Спирометрия.
Вследствие выраженной мышечной слабости дыхательной мускулатуры при измерении функции внешнего дыхания отмечаются рестриктивные нарушения в виде снижения жизненной емкости легких. - ЭНМГ.
При выполнении игольчатой электронейромиографии удается зафиксировать следующие изменения – резкое уменьшение скорости проведения и амплитуды вызванных потенциалов, спонтанную биоэлектрическую активность в покое (фасцикуляции, фибрилляции). - Гистология.
При патоморфологическом исследовании мышечного биоптата обнаруживается пучковая атрофия мышечных волокон, чередующаяся с неизмененной мышечной тканью, гипертрофированными миофибриллами и соединительнотканными разрастаниями. - ДНК-анализ.
Верифицирующий тест, позволяющий достоверно установить диагноз. Методом полимеразной цепной реакции выявляется генетическая мутация (делеция) SMN1 экзона 7.
Амиотрофию Верднига-Гоффмана следует дифференцировать с другими генетически обусловленными нервно-мышечными заболеваниями, имеющими такое же быстропрогрессирующее течение. К ним относятся врожденные структурные миопатии, ювенильный боковой амиотрофический склероз, синдром Фукуямы.
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар.
В тяжелых ситуациях (например, при выраженной гипоксемии вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ.
На сегодняшний день не существует этиотропной терапии амиотрофии Верднига-Гоффмана. Все мероприятия носят симптоматический и паллиативный характер. Применяются следующие методы лечения:
- Обеспечение питания.
Так как процесс глотания у многих пациентов затруднен, особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение ребенка – вертикальным. При необходимости устанавливается назогастральный зонд. - Физиотерапия.
Для улучшения метаболизма в мышечных тканях проводятся сеансы электрофореза, электростимуляция модулированным током, грязевые аппликации. - ЛФК.
С целью повышения мышечного тонуса необходимы регулярные физические упражнения – пассивные (выполняются специалистом) с постепенным переходом на активные (выполняются самим пациентом). - Ортопедическое лечение.
Для борьбы с костно-суставными деформациями, а также для их предупреждения используются ортопедические приспособления (корсеты, ортезы, иммобилизирующие шины), фиксирующие различные части тела. - Респираторная поддержка.
Важное место в лечении занимает устранение кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Медикаментозная терапия
Для достижения максимального эффекта лечение должно быть комплексным, проводиться непрерывно и подбираться индивидуально для конкретного пациента. Лекарственные препараты, применяющие для лечения амиотрофии Верднига-Гоффмана следующие:
- Метаболические средства.
Для улучшения метаболических процессов в нервных клетках и мышечной ткани назначается коэнзим Q10, L-карнитин, ноотропы. Также для стимуляции регенерации нервной ткани используют высокие дозы витаминов группы В (В1, В6, В12). - Вальпроаты.
Противоэпилептические препараты из группы производных вальпроевой кислоты способны увеличивать образование белка выживания мотонейронов (SMN), что впоследствии может улучшать клиническое течение заболевания. - ИПП и прокинетики.
Ингибиторы протонной помпы (пантопразол) и ЛС, ускоряющие моторику желудочно-кишечного тракта (итоприд), помогают в борьбе с гастроэзофагеальным рефлюксом, который часто возникает у больных АВГ вследствие выраженного нарушения глотания. - Муколитики и отхаркивающие средства.
С целью борьбы с такими дыхательными проблемами как слабое откашливание, скопление в дыхательных путях густой мокроты, применяются препараты разжижающие мокроту (ацетилцистеин) и стимулирующие ее отхаркивание (терпингидрат).
Хирургическое лечение
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. У лежачих больных, страдающих постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Болезнь Верднига-Хоффмана. Причины
Все формы болезни Верднига-Хоффмана вызываются мутациями в гене SMN1, расположенном в локусе 5q11-q13. Второй ген, известный как SMN2, также играет определенную роль в развитии этого заболевания. Ген SMN2 находится рядом с геном SMN1 на хромосоме 5. В то время как мутации в гене SMN1 являются причиной развития болезни Верднига-Хоффмана, некоторые группы исследователей получили доказательства того, что аномалии гена SMN2 могут влиять на тяжесть заболевания. У лиц с большим количеством копий гена SMN2, как правило, отмечается мягкая форма болезни Верднига-Хоффмана.
По всей видимости гены SMN1 и SMN2 кодируют белки, которые необходимы для правильного функционирования моторных нейронов. Некоторые исследователи считают, что ген SMN2 производит частично эффективный белок, необходимый для правильного функционирования двигательных нейронов. Вот почему люди с большим количеством копий гена SMN2 имеют мягкую форму болезни Верднига-Хоффмана.
Дополнительные гены также могут влиять на развитие этой болезни. Большее количество пациентов с болезнью Верднига-Гоффмана имеют делеции в гене NAIP. Некоторые исследователи предполагают, что потеря гена NAIP и / или различные мутации генов SMN могут играть определенную роль в тяжести заболевания.
Что происходит при заболевании
При исследовании пациента с этой патологией отмечается некоторое уменьшение спинного мозга в объёме. Ганглиозные клетки могут быть атрофированы, а могут совсем исчезнуть. В передних корешках выявляется дегенерация, снижение или полное отсутствие миелинизации в нервных волокнах, с отложением в них жировой ткани.
В мышцах скелета есть атрофированные пучки, которые переплетаются с нормальными волокнами, отмечается гиалиноз и сильное разрастание соединительной ткани, которая замещает мышечную ткань.
Болезнь Верднига—Гофмана имеет свою классификацию. По ней выделяют три стадии патологии.
- Врождённая, при которой симптомы начинают проявляться в первые несколько месяцев после рождения или сразу после появления на свет.
- Ранняя детская, при которой симптомы обнаруживаются в возрасте от 6 месяцев до 1,5 лет.
- Поздняя детская. Здесь основные признаки заболевания можно увидеть только после того, как ребёнку исполнится более, чем полтора года.
При этом самой тяжёлой формой считается именно врождённая.
Болезнь Верднига-Хоффмана. Лечение
Специфических методов лечения болезни Верднига-Хоффмана не существует. Все методы лечения направлены только на конкретные симптомы и проявления.
Гастростома может потребоваться детям, испытывающим проблемы при кормлении. Варианты контроля дыхательных проблем варьируются от использования неинвазивных процедур до долгосрочных инвазивных процедур, таких как трахеотомия. Любые решения по лечению должны приниматься только после консультаций всей медицинской бригады с родителями ребенка. Физическая и профессиональная терапии полезны для минимизации контрактур и для разработки компенсаторных стратегий. В некоторых случаях могут потребоваться ортопедические устройства (например, фигурные скобки) и хирургическая коррекция сколиоза.
Диагностика

Проявления проксимальной спинальной амиотрофии часто напоминают течение других неврологических и врожденных заболеваний, а также травматических повреждений структур спинного и головного мозга. Особенно затруднена диагностика этого заболевания у новорожденных и детей раннего возраста.
Ключевыми моментами в диагностике спинальной амиотрофии являются следующие исследования:
- Тщательный сбор анамнеза. Наличие случаев спинальной амиотрофии у родственников позволяет заподозрить это наследственное заболевание.
- Электронейромиография – специальное исследование нервно-мышечного аппарата. При этом исключается первичное поражение мышц и выявляются признаки, указывающие на патологию двигательных нейронов спинного мозга.
- Компьютерная и магнитно-резонансная томография. Эти методы позволяют иногда выявить атрофические изменения передних рогов спинного мозга. Однако, чаще они применяются для исключения другой патологии со стороны структур позвоночного столба и головного мозга.
- Биопсия мышц и с последующим гистологическим исследованием биоптата. Выявляются специфические изменения мышц, заключающиеся в чередовании пучковых атрофических и неизмененных мышечных волокон. Помимо этого, могут выявляться и компенсаторно гипертрофированные участки мышц, а также замещение мышечной ткани соединительной.
- Генетический анализ. Позволяет выявить точную причину заболевания: при исследовании ДНК выявляется мутация гена в пятой хромосоме.
Если в семье были случаи рождения детей со спинальной амиотрофией, при планировании последующей беременности супружеская пара направляется на консультацию к генетику. Также обязательной является дородовый анализ ДНК плода. Выявление синдрома Верднига-Гоффмана на стадии пренатальной диагностики служит показанием к прерыванию беременности.