Дистрофическая миотония (болезнь Штейнерта) выступает патологией генетического генеза с медленной прогрессией, основанная на наличии дефекта миотонин-протеинкиназы. Это приводит к формированию миотонии в комбинации с дистрофией миоволокон.
Болезнь Штейнерта передается потомкам по аутосомно-доминантному типу. Классический вариант формируется главным образом в возрасте от десяти до двадцати лет. И как редкость может развиться в первые дни жизни.
Морфологически наблюдается комбинация гипертрофических трансформаций одних миоволокон с атрофическими в иных, превращение некоторых в жировые и соединительно-тканные структуры. Гистологически происходит деструкция мышечных фибрилл и трансформация митохондриальной величины.
1.Общие сведения
Болезнь Лейдена-Томсена, или врожденная миотония, относится к группе наследственных болезней и встречается редко: распространенность в общей популяции оценивается как один случай примерно на 200 000 населения.
Наследуется врожденная миотония по аутосомно-доминантному типу, то есть для развития клинической формы достаточно получить дефектный ген от одного из родителей. В медицинскую нозологическую парадигму диагноз вошел с 1874-1876 гг, когда впервые были описаны семейные случаи заболевания.
Среди больных преобладают лица мужского пола.
Обязательно для ознакомления! Помощь в лечении и госпитализации!
Диагностические критерии
Подозрение на болезнь Россолимо-Штейнерта — Куршмана может возникнуть у врача при наличии у пациента сочетания миотонических и дистрофических изменений в мышцах на фоне потери интеллекта и наличия сердечнососудистой и эндокринной патологии.
Полисистемность практически всегда свидетельствует о генетической природе заболевания. Такие больные подлежат анализу ДНК и проведению генеалогического анализа для подтверждения аутосомно-доминантного наследования патологии. В качестве информативных методов исследования используются электрокардиография, электронейромиография, анализы на гормоны.
В связи с многогранностью клинических проявлений к процессу постановки диагноза обычно привлекаются специалисты из разных отраслей медицины — генетики, кардиологии, эндокринологии, гинекологии, андрологии, неврологии.
Дифференциальный диагноз проводится между дистрофической миотонией и другими видами схожих заболеваний. В отличие от остальных для болезни Россолимо характерна мышечная атрофия. Нередко для подтверждения диагноза приходится прибегать к биопсии, чтобы определить уровень мышечного белка, который в тканях при данной патологии повышен.
Проводится также антенатальная диагностика методом исследования околоплодных вод.
3.Симптомы и диагностика
Как правило, болезнь Лейдена-Томсена манифестирует и поддается диагностической идентификации в допубертатном или раннем пубертатном возрасте. Специфическим симптомом выступает остаточное напряжение (спастический тонус) каких-либо мышечных групп, которые были напряжены произвольно и адекватно ситуации, после чего должны расслабиться – и не расслабляются. Чаще всего такие тонические мышечные спазмы наблюдаются в пальцах рук, кистях, ногах, круговых мышцах глаза, мастикаторных (жевательных) мышцах. Соответственно, больной подолгу, в течение десятков секунд не может, например, открыть глаза или рот, повернуть голову, пошевелить конечностями, раскрыть ладонь, изменить выражение лица и т.п.
Характерной особенностью является нередко наблюдаемое противоречие между атлетической гипертрофией, ригидностью, рельефностью спазмированных мышечных групп и реально сниженной мышечной силой.
Рефлексологическое исследование с помощью неврологического молотка легко выявляет тонико-спастические реакции пальцев, языка и т.п. Из инструментальных методов диагностики наиболее информативным является электромиография. Обязательно назначается консультация медицинского генетика, собирается и в сопоставлении с клинической картиной изучается подробный семейный анамнез.
О нашей клинике м. Чистые пруды Страница Мединтерком!
Симптоматическая картина
Классический вариант
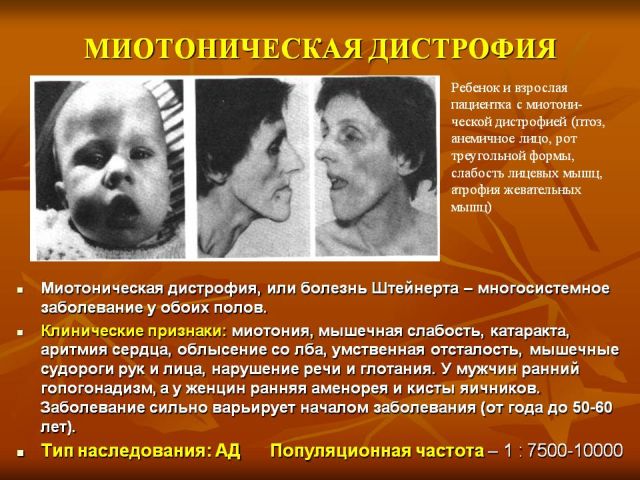
Проявляется после пятилетнего возраста и до 30 с лишним лет. Самый встречаемый возраст — от десяти до двадцати лет. Картина складывается из типичных признаков миотонического состояния с миопатией, повреждением сердечнососудистой системы и ЦНС, дисэндокринным состоянием и катарактой.
Из миотонических симптомов выступают спазмирования, особенно в жевательных миогруппах и мышцах, ответственных за сгибание кисти. Отмечаются и механические ответы, которые определяются во время постукивания нейрологическим молоточком. Характерным отличием выступают атрофические трансформации в разных мышечных группах. При таком состоянии течению патологического процесса характерно медленное уменьшение признаков миотонии на фоне прогрессирования миодистрофии.
Зачастую повреждаются миоволокна отдаленных участков конечностей, мимические, грудинно-ключично-сосцевидные и височные. Вовлечение в патпроцесс мимических мышц характеризуется возникновением маскоподобного печального лица заболевших. Атрофическая трансформация глоточной и гортанной миогруппы ведут к формированию миопатического пареза гортани с голосовой дисфункцией и трудностями при глотании. Патологические трансформации иногда формируются в мускулатуре респираторной системы. В комбинации со спазмированием они ведут к расстройству легочного вентилирования, развитию апноэ на фоне сна, пневмонии застойно-аспирационного генеза.
Дисфункция сосудисто-кардиальной системы отмечается в около 50% случаев:
- аритмия, которая связана с расстройством проводимости;
- гипертрофические явления левого желудочка;
- блокада ножек пучка Гиса.
Из проявлений повреждения ЦНС зачастую фиксируется гиперсомния и понижение интеллекта (вплоть до легкой степени дебилизма).
Эндокринные расстройства представлены главным образом со стороны половой системы. У лиц мужского пола представляются снижением либидо, крипторхизмом, импотенцией и пр., у лиц женского пола отмечается гирсутизм, нарушение цикличности менструации в виде олиго- либо дисменореи и ранний климакс. Характерным выступает трансформация строения волос в комбинации с аллопецией. Мужчины страдают выпадением волос на височной и лобной зоне, женщины – генерализованным либо очаговым облысением.
Врожденная форма
Начальные признаки иногда возникают еще при внутриутробном развитии в виде понижения мотоактивности плода, которое определяется по данным УЗ-исследования в третьем триместре беременности.
После рождения выступают признаки миопатии в виде диффузной миогипотонии, преимущественно в мимических, жевательных и глазо-двигательных мышцах и в отдаленных областей конечностей.
Этому состоянию свойственны трудности при вскармливании и диспное. Миотония появляется в более поздних периодах. Характерна олигофрения и задержка развития двигательной функции, быстрая прогрессия клинических признаков, которая зачастую оканчивается летальным исходом в раннем возрасте.
4.Лечение
Как и большинство других редких заболеваний, болезнь Лейдена-Томсена изучена недостаточно. Методы генной инженерии и генетической терапии находятся в стадии разработки и в клинической практике пока не применяются. Соответственно, терапия заболеваний этой группы всегда паллиативна и подбирается отчасти эмпирически, отчасти исходя из представлений о патогенезе. Для смягчения и редукции миотонических спазмов назначают диакарб, дифенин, соединения кальция (хлорид или глюконат) по определенной схеме. Достаточно хорошие результаты приносит физиотерапия и лечебная физкультура.
Мнения о «естественной» динамике врожденной миотонии Лейдена-Томсена в разных источниках расходятся: можно встретить сообщения как о неуклонном медленном прогрессировании в течение всей жизни, так и о тенденциях к спонтанному снижению выраженности спастико-миотонического синдрома.
В любом случае, болезнь Лейдена-Томсена никакой угрозы жизни не представляет, хотя, безусловно, в той или иной степени снижает качество жизни, вызывая психологический дискомфорт и очевидные трудности в социальной коммуникации (что является особенно болезненной проблемой на манифестном этапе, т.е. в подростковом возрасте). Поэтому важно обратиться за помощью своевременно, что позволит диагностировать заболевание и назначить адекватное лечение.
Причины развития миотонии Россолимо-Штейнерта-Куршмана
Миотония Россолимо-Штейнерта-Куршмана — это всегда генетически передающийся порок. Ключевой причиной его развития считается нарушение в гене DMPK, размещенном в девятнадцатой хромосоме. Тяжесть течения заболевания прямо пропорциональна количеству тринуклеотидных CTG. В норме этот показатель приблизительно равен 5-37, при 50-80 наблюдается мягкая форма заболевания, при 100-500 — поздняя тяжелая форма.
Под влиянием нарушения DMPK-гена в организме происходит изменение миотонинпротеинкиназы — белка, локализующегося в скелетной и мышечной ткани, в миокарде, ЦНС и др., что провоцирует появление миотонических спазмов, сочетающихся с атрофией мышц лица, шеи и конечностей в дистальных зонах. По сути, происходит гипертрофия одних волокон мышц с одновременной атрофией других. В результате часть этих волокон замещается либо другими волокнами, либо жировыми или соединительными тканями.
Возможные осложнения
Длительное течение заболевания приводит к вовлечению в процесс многих органов и систем.
Их поражение происходит опосредованно – вследствие длительного спазма мышц:
- Искривление позвоночника: вперед (лордоз), назад (кифоз), в бок (сколиоз);
- Межпозвоночные грыжи;
- Синдром ночного апноэ (удушье во время сна);
- Кардиомиопатия (поражение сердца развивается потому, что миокард представлен поперечно-полосатой мускулатурой);
- Мышечная атрофия;
- Стойкие изменения мимики, жевания и глотания;
- Бруксизм.
СПРАВКА. Курсовая терапия позволяет избежать осложнений и достичь длительной ремиссии, однако у пациентов часто развивается привыкание к противосудорожному средству. В связи с этим возможность самолечения и длительного приема одного и того же препарата без контроля врача должна быть исключена.
Симптомы миотонии Россолимо-Штейнерта-Куршмана врожденного типа
Врожденный тип миотонии может быть заметен специалистом еще в период пребывания в материнской утробе. Такой плод не проявляет повышенной двигательной активности. Поэтому если беременная женщина замечает подобное поведение малыша в животе, ей следует в обязательном порядке пройти УЗИ в последние три месяца перед родами.
Признаки миотонии присутствуют и у новорожденных. Это гипотония мимических, жевательных мышц, дистальных зон конечностей, глазных яблок. Кроме того, нередки и расстройства дыхания. Немного позже проявляются такие симптомы, как олигофрения и задержка моторного развития. Прогрессирование этих патологий может происходить с разной скоростью. При быстрых темпах их развития смерть пациента от заболевания может постичь его уже в самом раннем возрасте.
Дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршмана
Необходимо дифференцировать миотонию Россолимо-Штейнерта-Куршмана от прочих известных видов миотонии, которые в ряде ситуаций могут иметь схожие признаки и течение. Атрофия мышечных тканей характерна исключительно для этого заболевания и позволяет быстро отличить его от миотонии Томсена, признаком которой является гипертрофия мышц. Раннее поражение лицевых мышц и доминантное наследование отличают его от миотонии Беккера. Помимо указанных состояний, необходимо дифференцировать заболевание Россолимо-Штейнерта-Куршмана от БАС и амиотрофии Шарко-Мари-Тута.
Этиология и патогенез
Развитие болезни связано с генетической мутацией, развивающейся во время внутриутробного периода. Поврежденный ген, ответственный за работу хлорных каналов и синтез белка дистрофина, расположен на длинном плече 7 хромосомы. В результате мутации происходит недостаточное образование или полное отсутствие белка, регулирующего сокращение мышц.
В норме дистрофин отвечает за поддержание мышечной работы и правильную последовательность расслабления и сокращения. Из-за нарушения его синтеза происходит задержка хлора на мембранах мышечных клеток. Пучки поперечно-полосатой мускулатуры теряют способность к расслаблению и остаются сокращенными после выполнения движения.
Перенапряжение волокон вызывает еще большее накопление ионов хлора на поверхности клеточных мембран. Нарушается баланс между остальными ионными каналами. Накопленный хлор провоцирует выброс вне клеток ионов кальция. Избыток кальция оказывает прямое повреждающее влияние и приводит к разрушению мышечных клеток.
СПРАВКА. Тонические спазмы постепенно вызывают деформацию всего осевого скелета. Из-за повреждения дыхательных мышц развиваются эпизоды одышки или удушья. Повреждение поперечно-полосатой мускулатуры сердца приводит к кардиомиопатиям.
Лечение миотонии Россолимо-Штейнерта-Куршмана
В современной медицинской практике в настоящее время нет терапии, предназначенной для полного излечения людей от миотонии Россолимо-Штейнерта-Куршмана. Пациентам, у которых обнаружено это заболевание, назначают диету, основу которой составляют продукты с минимальным уровнем содержания калия или вовсе без него. Людям, страдающим данной миотонией, не следует переохлаждаться, так как низкие температуры могут в любом момент спровоцировать спазмы.
Для уменьшения риска их внезапного появления и улучшения общего состояния больного, ему назначают следующие препараты: хинин, новокаинамид, дифенин одновременно с диакарбом. К этим медикаментам специалисты рекомендуют добавить обязательный прием анаболических стероидов: неробола, ретаболила, метиландростендиола. Необходимо также включить в список обязательных для приема веществ небольшие дозы АТФ и витамины группы В.
Указанные выше препараты оказывают хороший эффект как при классическом, так и при врожденном типе заболевания. Все перечисленные меры позволяют снизить негативные влияния заболевания на организм пациента и продлить срок его жизни, однако они никак не способны полностью избавить его от миотонии и снова сделать абсолютно здоровым человеком.