Adolescent (juvenile) epilepsy first makes itself felt at the age of 10-12 years. The disease is divided into several types depending on the nature of its manifestations. More often, patients are diagnosed with a form of juvenile myoclonic epilepsy (Jantz syndrome). Generalized seizures are less common. Despite the characteristics of the disease in adolescence, there is no reason to limit the behavior of a teenager. Regular taking of medications ensures a full life.
Epileptic seizures
Almost all people imagine epileptic seizures in the form of a patient falling to the ground with loss of consciousness, as well as multiple very strong contractions of all the muscles of his body with foam at the mouth. However, this is only one way of manifestation of convulsive syndrome from a huge number of varieties.
The number of types of manifestations of seizures is very large; they can differ both in the patient’s sensations, preservation or loss of consciousness, the strength of the seizures, and in the area of localization.
Epileptic seizures can be presented in the form of spasms of any one muscle or their groups, spasms of one side of the body, and even muscles of internal organs, which is quite dangerous. Roughly speaking, almost all seizures, except those caused by a local effect on the muscles, can be called epileptic seizures of varying benignity or a seizure can be described by the predominance of signals of excitation of motor centers over signals of their inhibition.
The entire group of seizures is divided into clonic convulsions - prolonged muscle contraction and tonic - rhythmic contractions of muscle fibers.
Pathogenesis
Idiopathic variants of ME develop due to genetically determined increased excitability of cerebral neurons, leading to epileptogenic brain activity. Symptomatic myoclonic epilepsy is formed as a result of metabolic disorders and the accumulation of pathological compounds (polysaccharide inclusions, prion proteins) in nerve cells.
READ MORE: Pulmonary tuberculosis in adults: symptoms and treatment

In Lafora's disease, myoclonic encephalopathy of infants, increased epiactivity is due to dysmetabolism of neurons in conditions of proliferation of glial elements (with the death of neurons, impaired apoptosis of astrocytes). Neuronal hyperexcitability causes the occurrence of pathological nerve impulses going to muscle fibers.
The result is individual muscle contractions (myoclonus), tonic, clonic convulsions. Different localization of myoclonus reflects local excitation of different areas of the cerebral cortex. With diffuse spread of hyperexcitation, clonic-tonic paroxysm occurs with total involvement of muscle groups.
What is myoclonic epilepsy
Juvenile myoclonic epilepsy or juvenile (abbreviated JME), which is also called Janz syndrome, is a type of age-related benign epilepsy that is characteristic of adolescence and occurs during moments of restructuring of the body.

The likelihood of this disorder occurring is present in people from 8 to 26 years of age, but the vast majority experience attacks at 12-15 years of age. This is a very common syndrome, affecting a large number of adolescents, and very rarely young children.
General information
Myoclonic epilepsy (juvenile or juvenile epilepsy) is a benign disease that in most cases affects adolescents, which is why this disease is sometimes called adolescent.
The difference between a benign disease and a malignant one lies in the causes of its occurrence. Thus, a malignant disease occurs as a result of disorders in the brain associated with infectious diseases, tumors (heredity often plays a significant role in the development of attacks), etc.
Juvenile myoclonic epilepsy was first mentioned in 1867, however, only in 1955 in Germany, doctor Janz and his colleagues succeeded in identifying this ailment as a separate disease. Since that time, one could come across such a name for the disease as Janz syndrome.
This disease is diagnosed in 10% of patients diagnosed with epilepsy. The risk group includes adolescents aged 8 to 24 years. With the onset of puberty (puberty), the disease recedes and no longer bothers the patient. Despite this, a patient who has suffered from this disease should be monitored by a doctor.
Myoclonic spasms (myoclonus) are involuntary contractions of a single muscle/muscle group. Accordingly, epilepsy with a predominance of myoclonus in the clinical picture is called myoclonic. The concept of “myoclonic epilepsy” (ME) includes a number of diseases that are heterogeneous in etiopathogenesis, age of onset, and symptomatology.
In the vast majority of cases, they are characterized by a combination of myoclonus and generalized tonic-clonic seizures and are genetically determined. The incidence of ME varies; some nosological forms are so rare that no more than 100 clinical cases are described in the neurological literature.
Myoclonic epilepsy
Symptoms
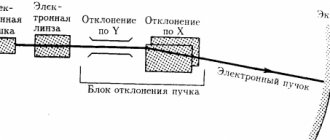
Myoclonic epilepsy in children manifests itself as attacks of generalized idiopathic seizures; any attacks are accompanied by a certain picture on the EEG, which is unlikely to be clear to a non-specialist.
The seizures are bilateral in nature, which, translated into simple language, means that the seizures involve paired organs: legs, arms, eyes, etc.
In this case, more often, convulsive activity occurs in the morning immediately after waking up, when the child has just gotten out of bed, and grabs his hands.
There are three types of JME attacks:
- Myoclonic - muscle twitching in certain areas of the body or the whole body, occurring mainly after physical exertion. In this case, consciousness is not lost, but the person cannot hold anything in his hands and may lose the ability to stand, feeling something like a blow to the knees.
- Tonic-clonic seizures are characteristic of 2/3 patients with myoclonic epilepsy and can occur as clonic or tonic convulsions with loss of consciousness and a feeling of suffocation.
- Absence seizures are a short-term loss of consciousness for a few seconds without convulsions.
Three stages of flow
Regardless of the form, the disease develops in three stages.
The first - epileptic-titaniform - lasts for several years, during which epileptic seizures develop and progress.
Additional symptoms are mental disorders, in which the patient often experiences unreasonable fear, attacks may be accompanied by painful spasms, and the person experiences increased sweating. Seizures always pass without loss of consciousness.
The second period is myoclonic-epileptic. Its development also takes years, and attacks, although they do not have a significant negative impact on life, spread not only to the facial muscles, but also to other parts of the body, affecting entire groups of muscle fibers.
In the severe stage of development of this stage, a person loses the ability to serve himself independently, and orientation in space is disrupted. Epilepsy attacks can be triggered by any sudden sound or flash of light.
The last, terminal stage, is characterized by an intensification of the described symptoms, but at the same time the patient experiences serious mental disorders.
Usually cognitive functions are not completely lost, but the person clearly has memory and speech disorders, and sometimes such people can be mistaken for the mentally retarded.
Causes
Juvenile myoclonic epilepsy, like any generalized idiopathic seizures, is hereditary in nature and is the result of a gene defect in the sixth human chromosome.

The threshold for seizure activity is also hereditary, so there is a theory supported by scientific research about a genetic predisposition to epileptic seizures.
Juvenile epilepsy is caused, like all benign seizures, by developmental features. When a child's body is rebuilt.
Benign JME develops from damage to brain tissue by any negative factors, and it is not true benign juvenile myoclonic epilepsy, but another type of epilepsy with the same symptoms.
JME attacks can occur either spontaneously or be provoked by the following factors:
- Lack of sleep.
- Overwork of any nature.
- Use of alcohol or psychotropic substances.
- Overexcitement.
Forms and stages of myoclonus
The origin of the disease cannot always be determined.
The main symptom of DMEM is seizure-like contractions of the muscles of the neck, limbs and head. Such seizures occur in children aged six months to three years two to three times a day and last no more than three seconds, so it is not always possible to pay attention to the presence of the disease in a timely manner.
This form is the easiest to treat, but if completely absent, complications may arise in the form of a lag in the development of psychomotor functions.
This pathology is also called “Dravet syndrome” after the French doctor Charlotte Dravet.
It is generally accepted that pathology manifests itself against the background of inflammatory processes in the central nervous system, which leads to disruption of the conduction of brain impulses, hence epileptic seizures.
Signs of the disease become apparent before the age of one year.
A predominantly hereditary form, which manifests itself in rapidly progressing and more frequent epileptic seizures (they mainly develop when exposed to external stimuli).
The disease is difficult to treat and symptomatic therapy is mainly used, which, with a successful combination of circumstances, can almost completely slow down the development of the pathology, but its final elimination is impossible.
Symptoms of the disease appear in the period from 5 to 16 years or later, and at first there are no epileptic seizures as such: the child only experiences twitching of the facial muscles, which is often mistakenly perceived as a nervous tic.
With age, twitching moves to the muscles of the shoulder girdle and upper limbs, and such attacks can be triggered by such external factors as sharp flashes of light, stressful situations and sudden excessive physical exertion.
The disease is also referred to by the acronym MERRF (myoclonic epilepsy, ragged-red fibers).
If other forms were described mainly at the end of the 19th century, then this type of pathology is relatively “new”: it was first officially recognized in 1980.
The disease occurs as a consequence of mutations in lysine transfer RNA, transmitted through the maternal line.
With such a violation, transfer RNA transmits inaccurate information inside cells, which leads to disruption of cellular activity, and this, in turn, is the cause of such neurological damage as epilepsy.
MERRF disease is the only one in the group that can manifest itself not only in children, but also in adults until old age.
The pathology rapidly progresses and manifests itself in the form of epileptic seizures and convulsions, dementia, deterioration of hearing and vision over the years, but timely symptomatic treatment gives positive results in most cases.
Diagnostics
Diagnosis of this syndrome is carried out using an electroencephalogram (EEG), which detects foci of increased neural excitation - foci of convulsive activity.
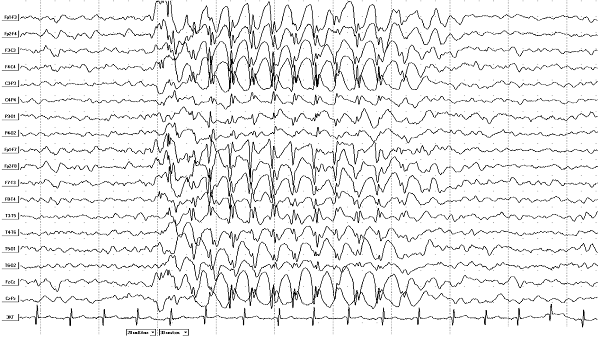
To exclude negative causes of the syndrome, a general blood test and brain tomography are additionally performed to find other diseases that can provoke epilepsy or confirm its benign nature.
Treatment regimen
The effectiveness of myoclinic epilepsy is currently low, and even in the case of successful selection of medications, the best outcome will be relief of symptoms, but not complete elimination of the problem.
Drug treatment is the main, but not the only form of treatment. In this case, the most effective use of anticonvulsants is:
- Clonazepam;
- Phenobarbital;
- Convulex;
- Benzonal;
- Depakine.
Typically, such treatment lasts at least two years, and given that all drugs used have a toxicity level at least above average, patients and their relatives are warned in advance about possible complications and side effects.
Due to such risks, doctors never focus solely on drug treatment, and in addition to eliminating seizures, family and social therapy are carried out, and the waking and sleeping patterns are adjusted.
Often the patient needs to eliminate the physiological causes of seizures (including removal of tumors in the brain: in approximately 10% of cases, the disease occurs against the background of the appearance of such tumors).
Features of helping the patient
Treatment of this disease is carried out only with medication. In some cases, the patient is prescribed surgical intervention. If the diagnosis was made correctly, then recovery occurs quite quickly.
Features of drug effects
A patient with juvenile myoclonic epilepsy is prescribed basic medications. The prescribed drugs are based on valproic acid derivatives.
If treatment does not bring the desired result, the doctor makes a decision regarding the prescription of polytherapy.
In this case, the patient is prescribed medications such as:
- suxilep and valproate (relevant in the presence of resistant absence seizures);
- phenobarbital and valproate;
- clonazepam, lamotrigine and valproate (relevant when myoclonic seizures are combined with photosensitivity).
In 75 percent of patients, absolute drug remission is observed. Most often, there is no need for polytherapy, and the result is achieved thanks to the effects of valproate. In 25 percent of patients, unfortunately, relapses are often diagnosed.
Cancellation of drug therapy is relevant in the 4th year of treatment. Even if the condition is relatively stabilized, drug treatment should not be stopped earlier.
Important to consider
Normalization of the daily routine is of great importance. First of all, this concerns sleep patterns. A person should train himself to go to bed and wake up at approximately the same time. Otherwise, the condition may be aggravated by insomnia.
It is equally important to constantly monitor the quality of your sleep. Considering that one of the main provoking factors is photostimulation, the patient should avoid bright light in every possible way.
So, you should not attend concerts of pop and rock performers. Until complete recovery, you do not need to visit bars and discos. You should also refrain from watching cartoons, anime and manga.
Types of attacks and accompanying symptoms

There are three types of myoclonus epilepsy attacks:
- Myoclonic. Characterized by muscle twitching that cannot be controlled. They usually occur in the morning, but can also be triggered by fatigue, strong emotions, alcohol consumption, sound or light exposure. The presence of cramps can be observed both in an individual muscle or limb, and throughout the body.
- Absence seizures. Consciousness turns off suddenly for a short period of time. It is caused by a lesion in the brain that affects its different parts. In this state, a person is in a stupor for several seconds, while his gaze also freezes, speech and movement stop. Despite their short duration, absence seizures lead to serious disturbances of consciousness.
- Tonic-clonic. The whole body is subjected to a convulsive state. In addition, loss of consciousness is observed. Involuntary urination or defecation is possible. The duration of the attack is several minutes. They are observed in more than half of patients and usually occur during awakening.
READ MORE: Numbness of the face with cervical osteochondrosis - causes of pathology, diagnosis, treatment methods and complications