Description
West syndrome.
Serial spastic contractions in individual muscle groups or of a generalized nature, occurring against the background of delayed neuropsychic development and accompanied by a hypsarrhythmic EEG pattern. Manifests before the age of 4 years, mainly in the 1st year of life. In most cases it is symptomatic. Diagnosis of the syndrome is based on clinical data and EEG results. To identify the underlying pathology, a CT or MRI, PET scan of the brain, and consultation with a geneticist or neurosurgeon are required. Treatment is possible with antiepileptic drugs, steroids (ACTH, prednisolone), vigabatrin. According to indications, the issue of surgical treatment is decided (callosotomy, removal of the pathological focus).
G40.5 Specific epileptic syndromes
Epilepsy (G40)
Excluded: Landau-Kleffner syndrome F Localized focal partial idiopathic epilepsy and epileptic syndromes with seizures with focal onset. Benign childhood epilepsy with EEG peaks in the central-temporal region. Childhood epilepsy with paroxysmal EEG activity in the occipital region.
Localized focal partial symptomatic epilepsy and epileptic syndromes with simple partial seizures. Seizures without changes in consciousness Simple partial seizures that develop into secondary generalized seizures.
Localized focal partial symptomatic epilepsy and epileptic syndromes with complex partial seizures.
Seizures with changes in consciousness, often with epileptic automatism. Complex partial seizures, turning into secondarily generalized seizures. Benign: Epilepsy with:. Partial continuous epilepsy [Kozhevnikova] Epileptic seizures associated with:. Epilepsy and epileptic syndromes not defined as focal or generalized.
Excludes: Landau-Kleffner syndrome F
Additional facts
West syndrome is named after the doctor who observed its manifestations in his child and first described it in 1841. Due to the manifestation of the syndrome at an early age and the occurrence of seizures as a series of individual spasms, the paroxysms that characterize West syndrome are called infantile spasms. Initially, the disease was classified as generalized epilepsy. In 1952, a specific hypsarrhythmic EEG pattern was studied, pathognomonic for this form of epilepsy and characterized by slow-wave asynchronous activity with random high-amplitude spikes. In 1964, specialists in the field of neurology identified West syndrome as a separate nosology. The introduction of neuroimaging into neurological practice has made it possible to determine the presence of focal lesions in the brain substance in patients. This forced neurologists to reconsider their views on West syndrome as a generalized epilepsy and classify it as a number of epileptic encephalopathies. In 1984, the evolution of the epileptic form of encephalopathy from its early variant to West syndrome, and over time to Lennox-Gastaut syndrome, was revealed. Currently, West syndrome accounts for about 2% of all cases of epilepsy in children and about a quarter of infantile epilepsy. The prevalence, according to various sources, is from 2 to 4.5 cases per 10 thousand newborns. Boys are slightly more likely to get sick (60%). 90% of cases of manifestation of the syndrome occur in the 1st year of life, with a peak at the age of 4 to 6 months. As a rule, by the age of 3 years, muscle spasms disappear or transform into other forms of epilepsy.
Classification
Depending on the nature of epileptic seizures, there are: 1. Focal epilepsy in children, occurring with focal (local, partial) seizures: • simple (with motor, autonomic, somatosensory, mental components). • complex (with impaired consciousness). • with secondary generalization (transitioning into generalized tonic-clonic seizures). 2. Generalized epilepsy in children, occurring with primary generalized seizures: • absence seizures (typical, atypical). • clonic seizures. • tonic-clonic seizures. • myoclonic seizures. • atonic seizures. 3. Epilepsy in children, occurring with unclassified seizures (repeated, random, reflex, status epilepticus, etc.). Localization-related and generalized forms of epilepsy in children, taking into account the etiology, are divided into idiopathic, symptomatic and cryptogenic. Among the idiopathic focal forms of the disease in children, the most common are benign rolandic epilepsy, epilepsy with occipital paroxysms, and reading epilepsy; Among the generalized idiopathic forms are benign convulsions of newborns, myoclonic and absence epilepsy of childhood and adolescence, etc.
Causes
In the vast majority of cases, West syndrome is symptomatic. It can occur as a result of intrauterine infections (cytomegaly, herpetic infection), postnatal encephalitis, fetal hypoxia, premature birth, intracranial birth trauma, asphyxia of the newborn, postnatal ischemia due to late clamping of the umbilical cord. West syndrome may be a consequence of abnormalities in the structure of the brain: septal dysplasia, hemimegaloencephaly, agenesis of the corpus callosum, etc. In some cases, infantile spasms are a symptom of phakomatoses (pigment incontinence syndrome, tuberous sclerosis, neurofibromatosis), point gene mutations or chromosomal aberrations (including Down syndrome). Cases of phenylketonuria with infantile spasms are mentioned in the literature. In 9-15%, West syndrome is idiopathic or cryptogenic, i.e., its underlying cause is not established or obvious. Often, the presence of cases of fibril convulsions or epileptic seizures in the family history of the sick child can be traced, i.e., there is a hereditary predisposition. A number of researchers indicate that vaccination, in particular the administration of DPT, may be a factor provoking West syndrome. This may be due to the coincidence of the timing of vaccination and the age of typical onset of the syndrome. However, reliable data confirming the provoking role of vaccines has not yet been obtained. The pathogenetic mechanisms of the occurrence of infantile spasms are the subject of study. There are several hypotheses. One of them associates West syndrome with a disorder of the functioning of serotonergic neurons. Indeed, patients experience a decrease in the level of serotonin and its metabolites. But it is not yet known whether it is primary or secondary. An immunological theory linking West syndrome to an increase in the number of activated B cells has also been discussed. The positive therapeutic effect of ACTH formed the basis of the hypothesis about malfunctions in the brain-adrenal system. Some researchers suggest that the syndrome is based on an excessive number (overexpression) of excitatory synapses and conducting collaterals, which form increased excitability of the cortex. They associate the asynchrony of the EEG pattern with a lack of myelin that is physiological for this age period. As the brain matures, its excitability decreases and myelination increases, which explains the further disappearance of paroxysms or their transformation into Lennox-Gastaut syndrome.
Reasons for development
Most often, the occurrence of symptomatic epileptic seizures is caused by the following provoking factors:
- volumetric formations of the central nervous system;
- hereditary metabolic diseases;
- alcohol withdrawal;
- infectious diseases of the nervous system (for example, meningoencephalitis);
- traumatic brain injuries (spontaneous or traumatic intracranial hematoma, brain contusions);
- prolonged insomnia and psychological stress;
- eclampsia;
- cerebral infarction, venous sinus thrombosis;
- hyperventilation, flickering light (if photosensitivity is present).
Symptoms
As a rule, West's symptom debuts in the first year of life. In some cases, its manifestation occurs at an older age, but not later than 4 years. The basis of the clinic is serial muscle spasms and impaired psychomotor development. The first paroxysms often appear against the background of a pre-existing delay in psychomotor development (PD), but in 1/3 of cases they occur in initially healthy children. Deviations in neuropsychological development are most often manifested by a decrease and loss of the grasping reflex, and axonal hypotonia. There may be a lack of eye tracking of objects and a disorder of gaze fixation, which is an unfavorable prognostic criterion. Muscle spasms are sudden, symmetrical and short-lived. Their serial nature is typical, with the interval between successive spasms lasting at least 1 minute. Usually there is an increase in the intensity of spasms at the beginning of the paroxysm and a decrease at the end. The number of spasms occurring per day varies from a few to hundreds. The most common occurrence of infantile spasms occurs during the period of falling asleep or immediately after sleep. Sharp loud sounds and tactile stimulation can provoke paroxysm. The semiotics of paroxysms that accompany West syndrome depends on which muscle group is contracting - extensor (extensor) or flexor (flexor). On this basis, spasms are classified into extensor, flexor and mixed. Most often, mixed spasms are observed, then flexion spasms, and most rarely – extension spasms. In most cases, one child experiences spasms of several types, and which particular spasm will predominate depends on the position of the body at the moment the paroxysm begins. A generalized contraction of all muscle groups may occur. But local spasms are more often observed. Thus, cramps in the neck flexors are accompanied by head nodding, spasms in the muscles of the shoulder girdle resemble a shrug. A typical paroxysm is a “jackknife” type, caused by contraction of the abdominal flexor muscles. In this case, the body seems to fold in half. Infantile spasms of the upper extremities are manifested by abduction and adduction of the arms to the body; from the outside it seems that the child is hugging himself. The combination of such spasms with a paroxysm of the “jackknife” type is associated with the “salaam” greeting accepted in the East, and therefore was called the “salaam attack.” In children who can walk, spasms can occur as drop attacks—unexpected falls while maintaining consciousness.
Treatment of episyndrome
If a person has an attack, then he needs help both at the time of the attack and in the future. Therefore, it is very important to know about the features of first aid and professional treatment of the syndrome.
First aid for an attack

If you have an attack, you should immediately call an ambulance
In the event of a seizure, the patient does not always need an ambulance. If the person is conscious, then he should be asked to sit down. If there is no deterioration in the condition, and the attack itself is short-lived, then you should not call a doctor.
If a person is unconscious, then he definitely needs help:
- You need to call an ambulance.
- The patient is placed on his back, his head is slightly raised. Folded clothing should be placed under the back of the head to prevent the person from hitting himself during convulsions.
- To prevent tongue biting, a folded handkerchief is placed in the patient's mouth. If a person's teeth are clenched tightly, it is better not to try to open them.
- If salivation is strong enough, then the patient's head is placed on the side so that he does not choke.
- Do not panic if breathing suddenly stops - it will resume after a few seconds.
Important! No attempt should be made to bring the person to consciousness.
The average duration of an attack is about three minutes. The main task of first aid is to prevent swallowing of saliva and tongue, and to prevent injury. If a couple of minutes after the attack a person feels better and wants to get up, you should persuade him to sit for 10 minutes and rest.
You should definitely call an ambulance if:
- a seizure occurred in a pregnant woman;
- the patient remains unconscious for 10 minutes;
- the attack lasts more than three minutes;
- the patient is a child or an elderly person.
Treatment options
If you have episyndrome, fried and smoked foods are contraindicated
Therapy is prescribed to the patient individually. This takes into account factors such as age and the degree of development of the underlying disease.
Treatment options for children and adults differ. Adults are usually prescribed:
- Anticonvulsants, which help reduce the frequency and intensity of seizures.
- Tranquilizers that help relax muscle fibers.
- Herbal medicine. Traditional medicine is an excellent addition to medications. They help relax muscles and improve the condition of the patient’s nervous system. The most commonly used decoctions are violet, linden, wild rosemary, celandine and oregano.
- Proper nutrition. Patients are advised to avoid smoked, fried and salty foods. You should also drink less fluid.
- In some cases, the patient is scheduled for surgery, which is performed by a neurosurgeon.
If an attack occurs in a child, treatment involves the following nuances:
- In case of high temperature, antipyretic drugs are prescribed.
- A diet that is low in protein and high in fat.
- Normalization of calcium-potassium balance.
- Phytotherapy.
It should be understood that attacks can be completely eliminated only by treating the root cause of their occurrence. Symptomatic therapy in this case is ineffective.
Differential diagnosis
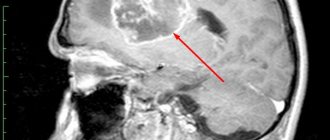
West syndrome should be differentiated from benign infant myoclonus, benign rolandic epilepsy, infant myoclonic epilepsy, Sandifer syndrome (head tilt like torticollis, gastroesophageal reflux, episodes of opisthotonus, which can be mistaken for spasms). Interictal (interictal) EEG is characterized by the presence of disorganized, chaotic, dynamically changing spike-wave activity, both during wakefulness and during sleep. Polysomnography can reveal the absence of spike activity during deep stages of sleep. Hypsarrhythmia is recorded in 66% of cases, usually in the early stages. Later, some organization of the chaotic EEG pattern is observed, and at the age of 2-4 years it transitions to the “acute-slow wave” complexes. The most common ictal EEG pattern (i.e., EEG rhythm during spasms) is generalized slow-wave complexes of high amplitude followed by suppression of activity for at least 1 sec. When recording focal changes on the EEG, one should think about the focal nature of the brain lesion or the presence of abnormalities in its structure. A CT scan of the brain in children with West syndrome may reveal diffuse or focal changes in cerebral structures, but may be within normal limits. In diagnosing local lesions, MRI of the brain is a more sensitive method. To identify areas of hypometabolism in brain tissue, in some cases it is possible to perform a PET scan of the brain.
G40 Epilepsy
Usually develops in childhood or young adulthood. Some forms of epilepsy run in families. Lifestyle and gender do not matter. In a patient with epilepsy, recurrent seizures or brief episodes of altered consciousness are caused by disturbances in the electrical activity of the brain. The condition usually develops in childhood, but the child may gradually outgrow it. However, older people can also develop epilepsy, since the likelihood of developing such disorders of brain function is high, for example, with strokes.
Most epileptics can lead normal lives. In 6 out of 10 patients with epilepsy, the cause of the disease remains unknown, although genetic factors may be involved. In epileptics, seizures can be triggered by lack of sleep or poor diet. Other triggers include alcohol abuse, flashing lights, and flickering TV or computer screens.
A single seizure is not always a symptom of epilepsy. For example, high fever in children can cause febrile seizures. Chronic alcoholics are also likely to have seizures both after binge drinking and as a result of complete abstinence from alcohol.
A sharp drop in blood glucose levels due to diabetes treatment can also cause a seizure in a diabetic. Epileptic seizures can be generalized or partial, depending on the degree of disruption of the electrical activity of the brain.
During a generalized or grand mal seizure, the functioning of the brain changes, while during a partial seizure, the activity of only one part of the brain is impaired.
Grand mal seizures are also divided into tonic epileptic seizures and absence seizures. Grand tonic epileptic seizure. It is preceded by a group of symptoms called an aura. The aura lasts a few seconds and gives the patient the opportunity to lie down or sit before completely losing consciousness and falling. In the first 30 seconds of a seizure, the patient’s body becomes very tense, breathing becomes intermittent and may even stop.
This stage lasts several minutes and may include uncontrolled movements of the limbs and trunk. After the seizure, consciousness returns to the patient, breathing becomes normal, and the muscles relax. Relaxation of the bladder muscles can cause urinary incontinence. The patient is confused and disoriented for several more hours, and may develop a headache. Status epilepticus is a severe condition in which the patient constantly has large tonic seizures, and between them the patient does not regain consciousness.
This condition can be life-threatening, and the patient should be under constant medical supervision. This form of seizures is also called petit mal seizures. The condition usually begins in childhood and can continue into adolescence. Absence seizures are rarely observed in adults. During a seizure, the child loses touch with the surrounding reality and falls into a trance with his eyes open. Each individual seizure lasts from 5 to 30 seconds, and after it the child does not remember what happened.
Because these seizures are never accompanied by falls or involuntary movements, they may not be noticed. Simple partial epileptic seizure. During this attack the patient remains conscious.
The head and eyes may be turned to one side, convulsions or twitching of one half of the body and face are possible, and the patient feels tingling sensations in these parts of the body.
After an attack, temporary weakness or paralysis of the affected part of the body occurs. The patient's perception of smells, sounds and taste changes. Psychomotor epileptic seizure. Before this attack, the patient’s perception of taste and smell changes, and there is also a feeling that all this happened once. He goes into a trance and does not respond to calls to him for several minutes. During an attack, the patient may smack his lips, grimace, and make small involuntary movements.
After a seizure, the patient does not remember what happened. Sometimes a grand mal seizure follows.
If possible, it makes sense to describe in detail the patient’s condition during the seizure in order to be able to give the doctor a complete picture of the patient’s condition. The doctor will do an examination to find the cause of the seizures, which may be a brain tumor or meningitis.
If the cause is not found and the seizures recur, the patient will have an EEG to look for abnormal brain activity. An EEG will also help diagnose a form of epilepsy because each form provides a different pattern of abnormal brain activity. In addition, CT scans and magnetic resonance imaging scans of the brain can be performed to look for possible structural abnormalities that cause epilepsy.
After a single seizure, treatment is usually not required. However, the underlying disease, such as diabetes, should be treated. If the patient has recurrent seizures, he will be prescribed anticonvulsants.
Doctors usually prescribe these medications in gradually increasing dosages until the seizures are controlled.
In some cases, a combination of anticonvulsants is necessary. The patient needs to have blood drawn regularly to monitor medication levels.
If he has not had seizures for 2-3 years, drug therapy is reduced or discontinued. However, any changes in the dosage of medications should only be done under medical supervision. Recurrence of seizures is possible within 2 years after discontinuation of medications. If medications do not control the seizures and the exact area of the brain responsible for the condition is found, surgery may be an option.
Patients with status epilepticus should be hospitalized immediately, as intravenous medications may be administered in the hospital to control seizures. If the patient has epilepsy, anything that could trigger a seizure should be avoided.
Provoking factors include lack of sleep, stress and flashing lights. If you witness an epileptic seizure, you need to turn the person on his side and protect him from involuntary movements. About 1 in 3 people who have one seizure will have another seizure within 2 years.
The risk of relapse is very high in the first weeks after the first seizure. However, for most patients with epilepsy the prognosis will be favorable, and more than 7 out of 10 patients will experience prolonged remission for 10 years. Makhiyanova and I. Maurer's original balm. Indomethacin Berlin-Chemie. Indomethacin 50 Berlin-Chemie. Royal capsules - vitamins and minerals.
Sodium para-aminosalicylate. Proserina tablets 0, Thyroliberin solution for injection. Ciprofloxacin hydrochloride. Risk factors The condition usually develops in childhood, but the child may gradually outgrow it. Diagnosis A single seizure is not always a symptom of epilepsy. Treatment After a single seizure, treatment is usually not required. Synonyms of the nosological group: Atypical convulsive seizures Atonic seizures Grand seizures Grand convulsive seizures Generalized absence seizures Jacksonian epilepsy Diffuse grand mal seizure Diencephalic epilepsy Cortical and non-convulsive forms of epilepsy Primary generalized seizures Primary generalized seizure Primary generalized convulsive seizure Primary generalized tonic-clonic seizure Pycnoleptic absence Repeated epileptic seizures Generalized seizure Convulsive seizure Refractory epilepsy in children Complex convulsive seizures Mixed seizures Mixed forms of epilepsy Convulsive state Convulsive seizures Convulsive states Convulsive forms of epilepsy Grand mal epilepsy Epileptic seizures Grand mal seizures in children.
Akatinol Memantine tablet. Amiridine 20 mg tablet. Amiridina tablets table. PE pack. PE 50 pack. Antigrippin-maximum caps. P syn. R red Apo-Fluoxetine caps. PE caps. PE 50 caps. PE 30 caps. PE 28 caps. PE 20 caps. PE 14 caps. Asentra tab. Maurer's balm original tincture, fl. Betamax tablet Van Bee caps. Vero-Ofloxacin tablet.
Treatment
West syndrome was considered resistant to therapy until the discovery in 1958 of the effect of ACTH drugs on attacks. Therapy with ACTH and prednisolone leads to a significant improvement or complete cessation of infantile spasms, which is accompanied by the disappearance of the hypsarrhythmic EEG pattern. Until now, there are no clear decisions among neurologists regarding the doses and duration of steroid therapy. Studies have shown that in 90% of cases therapeutic success was achieved with the use of large dosages of ACTH. The duration of therapy can vary between 2-6 weeks. A new stage in the treatment of infantile spasms began in 1990-1992. After discovering the positive therapeutic effect of vigabatrin. However, the benefit of treatment with vigabatrin has so far been proven only for patients with tuberous sclerosis. In other cases, studies have shown steroids to be more effective. On the other hand, steroid therapy has worse tolerability compared to vigabatrin and a higher relapse rate. Of the anticonvulsants, only nitrazepam and valproic acid have been shown to be effective. In some patients, the therapeutic effect of large doses of vitamin B6 has been described, which was noted in the first weeks of therapy. In case of infantile spasms, resistant to therapy, with the presence of a pathological focus confirmed on tomography, consultation with a neurosurgeon is indicated to resolve the issue of resection of the focus. If such an operation is impossible, then in the presence of drop attacks, a total callosotomy (intersection of the corpus callosum) is performed.
Forecast
Typically, by the age of 3, regression and disappearance of infantile spasms are observed. But in approximately 55-60% of cases they transform into another form of epilepsy, most often Lennox-Gastaut syndrome. Pharmacoresistance is often observed in infantile spasms accompanying Down syndrome. Even with successful relief of paroxysms, West syndrome has an unsatisfactory prognosis in terms of the child’s psychomotor development. Possible cognitive and behavioral disorders, cerebral palsy, autism, learning difficulties. Residual psychomotor deficit is not observed only in 5-12% of cases. Mental retardation is observed in 70-78% of children, movement disorders - in 50%. West syndrome, caused by abnormalities or degenerative changes in the brain, has a serious prognosis. In this case, the mortality rate can reach 25%. Cryptogenic and idiopathic West syndrome have a more favorable prognosis in the absence of mental retardation before the onset of spasms. In this group of patients, there is no residual intellectual or neurological deficit in 37-44% of children. Delaying the start of treatment has an adverse effect on the prognosis of the disease. Prognostic assessment is complicated by the fact that long-term consequences also depend on the underlying pathology against which symptomatic West syndrome occurs.