Prion diseases - transmissible spongiform encephalopathies (TSEs) - are a relatively new group of degenerative diseases of the central nervous system. The disorders are characterized by a chronic, unusual interaction between the immune system and the presumed causative factor, the prion protein.
In the peripheral path of the disease, it multiplies in the endoretular system, from where it passes into the central nervous system (brain and spinal cord), where it causes vascularization of neurocytes, glial cells, spongiomatous reconstruction of the neuropilus, extinction and reduction in the number of neuronal cells, gliosis without significant involvement of inflammation.
Prion - neuron killer
A prion, also known in microbiology as infectious PrP Sc, is a professional term describing a defective form of a protein commonly found in mammalian brain tissue that promotes long-term memory function.
Prions kill neurons very quickly, which provokes the rapid development of degenerative diseases.
According to the theory, we are talking about the source of many disorders of the central nervous system of animals (including humans). These proteins play a role in hereditary changes and evolution.
One of the regulatory factors of prion conversion is chaperones, whose function is to clean cells from pathogenic proteins. Both chaperones and prions belong to the group of cellular proteins.
Theories and hypotheses
The theory explaining why prions kill neurons was formulated by Professor S. B. Prusiner in 1982. He was also the first to use the word "prion" (originally intended to be used as a portmanteau of "proteinaceous" and "infectious", but it sounded better in the altered state).
Prusiner formulated the theory of prions as pathogenic proteins in connection with the search for the causative agent of Creutzfeldt-Jakob disorder. The agent he discovered was something new. Until 1982, it was believed that infectious diseases could only be caused by infectious organisms containing nucleic acid carrying genetic information. But the protein structure does not contain nucleic acid. It is a protein that multiplies by changing similar proteins in the body. In 1997, S. B. Prusiner received the Nobel Prize in this field.
Pathogenic prion proteins have the same primary structure (amino acid sequence), but differ in their conformational configuration. While wild PrP c has a strong α-helix predominance and about 5% β-sheet, pathogenic PrP Sc (Sc – scrapie) has a β-sheet proportion of up to 40%. The reasons for the aberrant properties of PrP Sc prions are still under investigation. Today there are several hypotheses.
- The viral hypothesis explains the effects of prions by virology. The involvement of RNA viruses in transmissible spongiform encephalopathies has been suggested. Both viroids and prions are small infectious pathogens; therefore, the result of exposure to the virus is a prion of an infectious nature.
- Multicomponent hypothesis . It is assumed that for the formation of infectious proteins as new pathogens, communication with polyanions and lipids is necessary.
- Heavy metal poisoning . Intoxication causes the development of infection when there is a lack or excess of copper in the body (an optimal amount of copper is necessary for healthy protein).
What is the toxicity of protein to a neuron?
Once the infectious protein appears, it can “imprint” its conformation onto healthy neurons. Toxic proteins can kill mitochondria in neurons, spreading disease to tissues.
Pathogenic proteins are extremely resistant to physical and chemical influences, which leads to difficulties in sterilization (attempts have been made to burn the brains of affected animals at 600°C; the ash subsequently infected approximately 1/3 of the animals). In accordance with what metabolic processes are disrupted by prions in the neuron, the most infectious are the tissues of the eye, brain and spinal cord.
Scisne?
Molecular biologist Byron Coie on the structure of prions, prion diseases, their causes and treatment.
Creutzfeldt-Jakob disease |
Prions constitute a separate class of infectious agents;
they are protein based and do not contain a genome consisting of nucleic acids. The concept of the existence of prions includes the idea of a new, protein heritability - altered forms of proteins in the host organism, which, when transferred to a new organism, can cause a change in phenotype in the recipient. Most prions are pathogens, but compared to other classes of pathogens (eg, viruses, bacteria, fungi, parasites) they are unique in that they spread within and between hosts without transferring or replicating their own DNA or RNA. Prions are typically refolded and aggregated proteins that spread by invading the host and stimulating the refolding of the corresponding normal form of the protein. The prion aggregate grows and then somehow fragments to generate more prion aggregates. In many ways, this is similar to Kurt Vonnegut's Ice-Nine: the growth of prions is similar to the growth of crystals, and prions themselves are often described as seeds, in an analogy to seed crystals. Thus, prions do not need to transfer their genetic code, but the host organism must create the normal protein that makes up pathogenic prions. Many (if not most) proteins can rearrange and/or assemble into ordered aggregates that can grow under certain natural or experimental conditions—in tissue or in vitro—but not all such protein aggregates are pathogenic prions. The term "prion" means that, at a biological level, the refolded protein structure can spread between hosts or at least from cell to cell within a multicellular host organism. Recently there has been a proposal to collect under the umbrella term "prions" all states of a protein that promote its growth in the form of multimeric aggregates, but this expanded use of the term violates the underlying idea of transmissibility and does not allow prions to be distinguished from the many other cellular structures that can grow. but do not tend to spread to other cells and organisms.
History of research
The prototype of prion disease was a transmissible neurodegenerative disease of sheep - the mysterious deadly scrapie (or sheep scrapie). Early research has shown that the scrapie agent is unusually resistant to treatments that neutralize other pathogens and can persist in pastures for years. The fact that scrapie exhibits resistance, in particular to radiation, led J. S. Griffith and Tikvah Alper to propose in the 1960s that it represents a new class of pathogens that does not have its own nucleic acid genome and can be an abnormal self-replicating form of a protein or membrane. Meanwhile, Carlton Gajdushek's descriptions of brain pathology caused by the human disease kuru in Papua New Guinea led William Hadlow to believe that kuru was similar to sheep scrapie, and Hadlow recommended that kuru be tested for transmission from humans to other primates. Gajdushek successfully carried out this work and showed that the Fore people fell ill with kuru during ritual cannibalistic holidays. A striking feature of kuru and other prion diseases that often obscures their causes is the long incubation period between infection and the appearance of clinical signs, which in humans can exceed four decades.
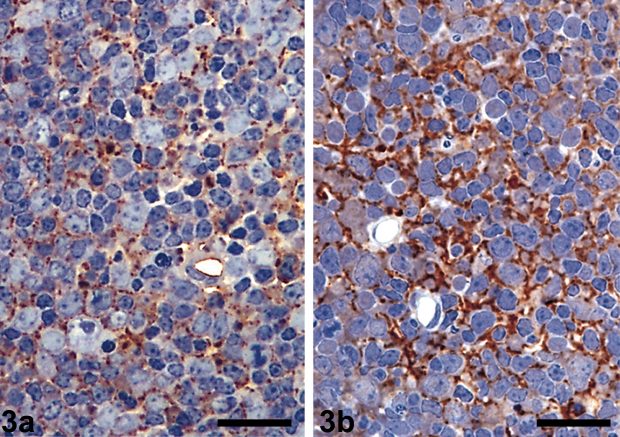
Lymph nodes from healthy (a) and infected (b) sheep - body staining clearly shows evidence of scrapie disease prions in tissue from infected sheep / wikipedia.org |
In the 1980s, Stanley Prusiner coined the term “prion” for these agents and was the first to identify the specific protein that is the main component of scrapie prions, the prion protein (PrP).
Homologues of the same protein have been found in many mammalian species, including humans, and abnormal PrP aggregates have been identified in other transmissible neurodegenerative diseases of humans and animals, like scrapie. These diseases are now known as prion diseases, or transmissible spongiform encephalopathies. Stanley Prusiner, Charles Weissman, and other researchers have shown that PrP is an important susceptibility factor for prion diseases. My laboratory, in collaboration with Peter Lansbury, has shown that disease-associated forms of PrP can themselves cause the transformation of normal PrP molecules into abnormal forms. In these conversion reactions, we identified striking biochemical features that help explain the characteristics of known prion strains and the barriers to their transmission between different species. However, to unequivocally prove that prions consist of refolded PrP aggregates and do not require prion-specific nucleic acids, it was necessary to develop methods for continuous cell-free prion amplification or de novo prion formation reactions. They were originally created by the laboratories of Soto, Supattapone and Prusiner in the 2000s; Until then, it had been difficult to completely rule out the possibility that these diseases were caused by unidentified viruses.
Although the word "prion" was first applied to the transmissible spongiform encephalopathies described above, the first clear evidence that infectious proteins exist in biology came when Reed Wickner realized in 1994 that some unexplained epigenetic elements in yeast were prions. These prions did not consist of a homolog of PrP, but of very special yeast proteins. The relative simplicity and power of yeast biology and genetics allowed Wickner and other researchers to clearly demonstrate a number of fundamental principles of prion biology and structure that had been much more difficult to identify in mammalian prions.
Research methods
Unfortunately, many of the standard techniques that have long guided the study of common pathogens—pathogen genetics, serology, X-ray diffraction, nuclear magnetic resonance (NMR) spectroscopy—are extremely difficult to apply to prions. Without any specific pathogenic genes that can be sequenced or mutated, many standard genetic and reverse genetic approaches to identifying the structure and function of pathogens do not work. Because prions are composed of host proteins, there is very little host immune response to the pathogen; thus, simple serological detection of prion infections based on antibody interactions is very difficult. In addition, mammalian prions tend to be tightly packed, highly glycosylated, and associated with other host molecules, and therefore even specific prion conformational epitopes (surfaces recognized by antibodies) on PrP aggregates are difficult to detect and exploit. All attempts to determine the three-dimensional structures of prions have been stalled for a long time, since purified prions have an aggregated, but non-crystalline nature.
For many years, the only way to detect and analyze mammalian prions was through animal bioassays, which even in the fastest models, rodents, took several months to one year. Within a given organism, different strains can usually be distinguished by incubation periods, neuropathological patterns, and biochemical signatures of disease-associated PrP deposits or prions.
Fortunately, powerful cell-free prion amplification assays have recently been developed, such as prion protein cyclic amplification (PMCA), real-time vibration-induced conversion assay (RT-QuIC), and scrapie cell assay. These methods rely on the inherent replication mechanism of prions. Both PMCA and RT-QuIC are extremely sensitive: they can amplify the presence of prions by a factor of a trillion, almost to the point of detecting a few prion particles. PMCA reactions propagate prion infection, thereby reflecting and illuminating many aspects of prion biology, while RT-QuIC assays generally do not propagate infectious prions completely, but provide faster, more practical, and higher throughput methods for their detection, and, thus, they have become state-of-the-art tools in diagnosing prion diseases. Both PMCA and RT-QuIC are useful in some cases in distinguishing important prion strains in certain host species.
Progress has been slow in identifying the basic structure of prions. Semiconductor NMR studies have revealed the molecular architecture of some fungal prions and prion-like fibrillar structures of mammalian PrP. Electron crystallography, fiber diffraction, and cryoelectron microscopy studies have helped characterize the key structural limitations of mammalian prions, but the application of these and perhaps other structural biological techniques remains to be improved.
Structure and reproduction of prions
Understanding the structure and mechanisms of replication of mammalian prions, at least at the molecular level, is extremely difficult. We first need to explain how misfolded proteins can spread as pathogens without carrying their own nucleic acid genome. It should then also be explained how proteins with a single amino acid sequence, such as PrP from a given animal host, can form different strains of prions that propagate smoothly and cause different disease phenotypes without the genetic mutations that account for strain variation in common pathogens.
Many studies indicate that mammalian prions are ordered aggregations of several PrP molecules, tightly packed and often fibrillar or filamentous. PrP molecules (monomers) in prions are almost completely reversed compared to normal free PrP molecules. When the correct PrP molecules are incorporated into growing prion aggregates, these aggregates cause them to refold, with the prions acting as strain-specific templates or seeds that somehow impart their own aberrant forms to each incoming molecule, controlling the stable replication of their strain.
Beyond this rough description, details of the structure and distribution of prions at the molecular level remain unclear. Also unresolved is the question of how prions spread beyond the initial site of infection in the host. Existing evidence suggests that the most efficient cell-to-cell transmission of prions involves membranous structures such as exosomes or tunneling nanotubes, most likely because prions are typically associated with membranes by lipid anchors; however, the potential of these membrane structures to promote prion spread in vivo remains to be determined. Understanding the mechanisms of prion propagation is important because the ability of various misfolded protein aggregates to spread within and between cells, tissues, and individuals determines whether they act as infectious pathogens or are relatively harmless disruptions of protein metabolism.
Prion diseases
Many mammalian species, including humans, lesser primates, cattle, sheep, goats, deer, elk, cats, mink, rodents, and various exotic ungulates, are susceptible to PrP prion diseases. But not all species are this way: dogs and horses appear to be notable exceptions. Different species typically express slightly different normal PrP molecules, and differences in the amino acid sequence of PrP can greatly influence the host's susceptibility to incoming prion infections. For example, humans are known to be somewhat susceptible to bovine spongiform encephalopathy (BSE) but appear to be resistant to sheep scrapie and, to our knowledge, chronic wasting disease of deer. For some reason, forest voles and squirrel monkeys are unusually susceptible to a wide range of prion infections from other species.
The mechanisms by which prion infections cause neurodegenerative diseases are still unknown to us. Aggregates of various prion strains in host organisms of different species can accumulate predominantly in different areas of the central nervous system and cause a number of neuropathological disorders. It is clear that the end effect of at least partial damage is neuronal failure and loss, causing a variety of clinical symptoms and leading to death. A number of neurophysiological processes and pathways are known to be impaired, but much remains to be determined as to whether such impairments are associated with direct or indirect prion toxicity and to what extent one or another deficiency or combination of deficiencies is most responsible for the demise of the patient.
In humans, the causes of prion diseases may be genetic (due to specific mutations in the PrP gene), acquired (caused by contamination - such as exposure to kuru, BSE, or other prion-containing material), or sporadic (of unknown origin; usually thought to be due to spontaneous prion production for a specific individual). The vast majority of human prion diseases are sporadic, with sporadic Creutzfeldt-Jakob disease (sCJD) being the most common, with an annual incidence of approximately one per million population worldwide. A number of different mutations in the PrP gene can cause a variety of familial prion diseases, with some mutations being completely penetrant (always causing disease in carriers of the mutation) and others being less penetrant. Clinical symptoms and disease progression may vary markedly among different hosts and among different types of prion diseases, but may include dementia, incoordination, insomnia, hallucinations, muscle stiffness, confusion, fatigue, and difficulty speaking.
There are also important animal prion diseases. BSE emerged as a widespread cattle epidemic due to what might be called "agricultural cannibalism" in the 1990s. Consumption of BSE—contaminated beef—caused nearly two hundred cases of variant Creutzfeldt-Jakob disease in humans at the time, but preventative measures have nearly eliminated BSE and prevented new cases from occurring. Chronic wasting disease in deer is sweeping across North America at an alarming rate, with cases also emerging in South Korea and Norway. Scrapie is an ongoing problem with sheep and goats in many parts of the world.
Diagnosis and treatment of prion diseases
Recently, significant advances have been made to accurately and relatively non-invasively diagnose human prion diseases in living patients based on new prion-specific tests of nasal, cerebrospinal fluid, blood, urine or skin swabs. For example, RT-QuIC testing of cerebrospinal fluid and/or nasal brush biopsy materials can achieve 100% accuracy in diagnosing sporadic Creutzfeldt-Jakob disease. These tests are beneficial because they measure prion disease pathogens, but they are not yet fully validated or officially recommended by organizations such as the WHO. Otherwise, diagnosis of sporadic prion diseases in humans depends primarily on a combination of clinical signs, brain scans, electroencephalograms, and other biomarkers, which together may have high diagnostic sensitivity but are not entirely specific for prion diseases.
Despite the recent advances in the development of new prion tests described above, current guidelines are that definitive diagnosis of sporadic or acquired prion disease requires neuropathological examination of brain tissue obtained by biopsy (rarely) or autopsy. I believe that these recommendations will soon be changed to include new, less invasive intravital tests for detecting prions. Unfortunately, although this progress in the early diagnosis of prion diseases should improve the prospects for the development and use of therapeutics, there are currently no treatments available that have proven effective in clinical trials.
Open Issues and Future Research Directions
In the field of mammalian prion diseases, the following key questions remain: “What is the self-propagating structure of prions and how does it vary depending on the prion strain?”, “How does prion activity lead to brain damage?”, “How can we prevent this damage or repair it when treatment of prion diseases?”, “What are the most relevant mechanisms of transmission of prion diseases in humans and animals?”, “Which animal prion diseases (besides BSE), if any, have zoonotic potential, that is, can cause disease in humans?”, “ To what extent do other pathogenic, misfolded proteins, which can also act as seeds, behave like PrP-based prions in their ability to spread within or between individuals to cause disease?”
Indeed, the latter question represents an important milestone in the study of many diseases associated with protein misformation, especially those associated with the pathogenic accumulation of abnormal fibrillar protein deposits (eg, amyloid fibrils and plaques). These diseases include Alzheimer's, Parkinson's and Huntington's diseases, as well as amyotrophic lateral sclerosis, frontotemporal dementia, chronic traumatic encephalopathy and type 2 diabetes. Various host proteins form aggregates in these and many other diseases, but, like prions, such aggregates usually grow by seeding normal soluble protein molecules. Thus, the potential for prion-like propagation of proteins exists at the molecular level. There is also growing evidence that many different disease-associated protein deposits can grow and spread in the same way as prions, causing pathologies after inoculation into localized areas in experimental animals.
The results of these studies raise pressing questions about whether numerous protein refolding diseases—which are often much more common than PrP-based prion diseases—can be transmitted to humans or animals in the real world. Creutzfeldt-Jakob disease is transmitted between people through tissue transplants, injections of hormones obtained from corpses, blood transfusions and contaminated medical instruments. A second factor in such iatrogenic transmissions is the fact that prions are often not completely inactivated by standard clinical disinfection procedures.
It remains to be determined whether other types of potentially prion-like, disease-associated protein aggregates can be identified that may also be resistant to inactivation and yet capable of initiating or accelerating pathogenic processes in humans. I am not aware of any epidemiological indication that this is the case, but further careful study of this issue seems warranted.
Byron Coie - Ph.D., Chief of the TSE/Prion Biochemistry Section of the Laboratory of Persistent Viral Diseases, National Institute of Allergy and Infectious Diseases. PostScience
Risk factors for developing infections
A risk factor for diseases from a genetic point of view is the presence of a certain sequence of bases in a certain place in the gene encoding a protein.
A high risk of the formation of infectious prions and prion diseases is represented by operations with tissue contamination and transplantation from a patient with an existing infection.
An unproven risk is the consumption of tissue from a sick animal. The important factor here is the toxicity of the prion, its ability to survive when exposed to temperatures several times higher than the boiling point of water. Livestock farming without food consumption is not risky.
The most dangerous (infectious) from the affected organism are the brain (CNS), meninges, cornea (eye), blood, lymphatic organs. Muscles and other organs pose a much lower risk, but do not eliminate it.
Routes of infection
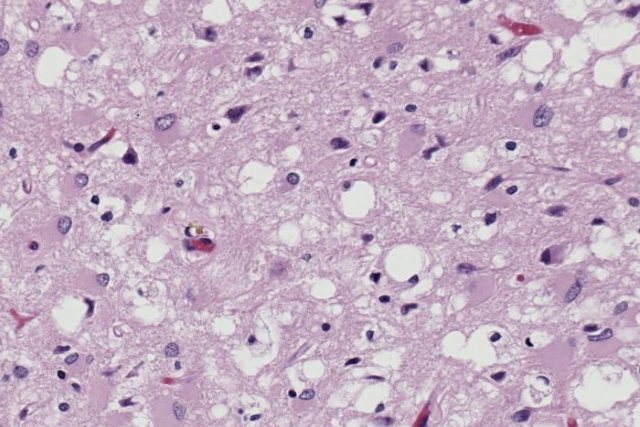
Histological specimen showing tissue damage by prions with the formation of a characteristic spongy structure.
https://www.youtube.com/watch?v=upload
Very little is known about the molecular nature of disease-causing prions. Infection can be caused by approximately 100,000 molecules, which in most cases form large clumps. The significance of the aggregation of individual molecules in association for the virulence of prions is not yet known. It cannot be ruled out that individual prion molecules are also virulent.
Some experiments suggest that for prions to appear in tissue, only temporary contact of the tissue with material containing prions is sufficient, and there is no need for prions to be permanently introduced into the body. This risk is relevant, for example, in connection with the use of surgical instruments contaminated with prions.
The course of the disease and the spread of prions throughout the body depends on the type of prion. Prions differ in the composition of amino acids characteristic of a given species, determined by the species prion protein gene, as well as so-called post-translational modifications or the degree of glycosylation of the basic protein chain.
Post-translational modification significantly influences the characteristics of prions and is responsible for the difference between the so-called prion genera. In the case of the new variant (nvCJD), only one type of prion has been described so far, similar to the prions of livestock infected with bovine spongiform encephalopathy. Therefore, the course of the disease in humans and animals infected with the new variants is almost the same.
In other species of living beings, however, many prion genera are known. About two dozen such genera have been described in sheep, which are not virulent for humans. The course of sheep prion disease varies dramatically depending on the type of prion - from very fast, with almost sudden death, to slow, protracted.
Atypical cases of the clinical course of the new variant in cattle infected with bovine spongiform encephalopathy, which occurred in Japan and Italy, suggest the existence of more genera of bovine prions. If this genus of bovine prions were to enter the human body, one would expect the emergence of a new variant with symptoms and a clinical course different from known cases.
In patients with Creutzfeldt-Jakob disease, prions spread to the nervous system, eye tissue, and lymphatic tissues, including the tonsils, spleen, and cecum. The largest number of prions is found in the nervous system, and the smallest in the lymphatic tissue.
So far, no cases of new variant Creutzfeldt-Jakob disease (nvCJD) transmission through medical intervention have been reported, which is, of course, good news. On the other hand, experts warn about exaggerated optimism, especially in the UK, since the incubation period can be quite long (from 5-8 months to 10-15 years).
Characteristics of infections
Diseases are caused by pathogens that do not contain nucleic acid. They can be sporadic, genetically determined, infectious (even iatrogenic). The presence of defective proteins causes disorders commonly called spongiform encephalopathies. These are degenerative disorders of the nervous system in which the brain gradually takes on a spongy appearance due to miniature holes due to prions killing neurons.
All infections are incurable and fatal. Today, research and testing are being conducted to explore methods of slowing the progression of prion disease.
Prions and medical instruments
Prions are very resistant to conventional disinfection methods. Ionizing, ultraviolet or microwave radiation has practically no effect on them. Disinfectants commonly used in medical practice have only a very limited effect on them. They are reliably eliminated by disinfecting reagents - strong oxidizing agents that have a destructive effect on proteins.
Another difficulty is the resistance of prions to high temperatures. Even with autoclaving at 134 °C for 18 minutes, complete destruction of prions cannot be achieved, and prions “survive” in a form that can cause infection. Resistance to high temperatures is further enhanced if the prions have dried on a metal or glass surface or if the samples have been exposed to formaldehyde before autoclaving.
In the UK, where the new variant is a very serious problem, disposable surgical instruments are already being used for tonsillectomies for these reasons. In the future, an alternative solution arises: the creation of new instruments, taking into account increased requirements for cleaning and disinfection.
A much more complex solution to this problem is the treatment of patients at risk. These include patients who have undergone operations that involve potentially infected dura mater, or patients from families with a hereditary form of Creutzfeldt-Jakob disease. WHO does not require any special measures in this case.
Individual nosological units
Human prion diseases are species specific. Basically, they spread only among individuals of one species. But scientists have demonstrated interspecies transmission (overcoming the interspecies barrier): this occurred in scrapie sheep with its transmission to cows (mad cow disease). Transmission of the disease from animals to humans, despite many suspicions, has not been demonstrated.
A number of diseases are of hereditary origin, some are infectious. Sometimes the disease occurs unexpectedly, without the possibility of prevention.
The disease existed only in Papua New Guinea in tribes of cannibals who consumed the brains of the dead. It manifests itself as tremor (“kuru” means “tremor”), speech disturbances, and balance disorders (ataxia). Death occurs a few months later. After stopping cannibalism, the kuru disease disappeared.
Creutzfeldt-Jakob disease
It is a rare disorder that affects older people (most often 50-70 year olds). Its course is relatively rapid, death occurs within a few months (up to a year) from the onset of the first symptoms. Neurons quickly die, and progressive dementia develops, which has no known cure. There is some hereditary predisposition to this disease:
- Sporadic form . The incidence is 1-2/1000000. Problems begin around age 65. The disorder occurs in the form of acute progression of dementia (over 2-3 months), ataxia, and myoclonus. There are no ways to prolong the life of neurons with this prion disease; the patient dies 5-12 months after the appearance of the first symptoms.
- Iatrogenic form . It occurred in patients receiving human growth hormone from cadaveric pituitary glands (today it is prepared recombinantly), transplantation of hard membranes, pericardium, and cornea. There is a risk of neurosurgical transmission. Infection is also possible through transfusion.
- Family form . This is a genetic form with a mutation in the PRNP gene and neuropsychiatric symptoms.
- New option . Characterized by psychiatric symptoms (anxiety, depression, behavioral changes), progressive cerebellar syndrome, myoclonus, and other neurological symptoms. Unlike the sporadic form, the new variant is characterized by a slow progression of viral infection; prion disease affects younger age groups. Transmission occurs through nutritional means (through consumption of meat from affected animals). The incubation period is more than 10 years. Around 200 people have died worldwide to date.
- The childhood version of Creutzfeldt-Jakob disease is Alper disease . Has the same manifestations as the disease in adults; in addition accompanied by hepatic steatosis.
Gerstmann-Sträussler-Scheinker disease
Gerstmann-Strüssler-Scheinker disease is a very rare autosomal dominant hereditary disease that begins in the 3rd-4th decade of life. It manifests itself as a slow slowdown in the functioning of neurons by prions, and progressive dysfunction of the cerebellum and spinal cord. Signs:
Properties of molecules
Three-dimensional structures of the C-terminal regions of PrP proteins
C
(left) and PrP
Sc
. Alpha helices are colored purple, beta strands are colored green.
The N-terminal region of the protein is relaxed and cannot be analyzed by X-ray diffraction.
https://www.youtube.com/watch?v=i1pt_
Mammalian prion proteins are not similar in amino acid sequence to yeast prion proteins. Despite this, the main structural features (formation of amyloid fibers and high specificity that prevents the transfer of prions from one type of organism to another) are common to them. However, the prion responsible for mad cow disease has the ability to be transmitted from species to species.
The right picture is a model of two prion conformations; on the left is the known, normal, structure conformation of the terminal region of the C-terminal PrPC. (see RCSB Protein Databank for display/download).
Animal diseases
The risk of animal diseases for humans is very speculative (the problem is best characterized by the connection between BSE and a new variant of Creutzfeldt-Jacobs disorder). In animals, disorders are manifested by aggressiveness and impaired motor abilities:
- bovine spongiform encephalopathy (BSE);
- scrapie;
- chronic wasting disease (CWD);
- felt spongiform encephalopathy;
- transmissible encephalopathy.
Research on yeast prions and other micromycetes
https://www.youtube.com/watch?v=ytcopyrighten-GB
Prion-like proteins, whose behavior is similar to that of PrP, are found in natural populations of micromycetes and yeast. Studies of yeast prions have supported the hypothesis that the conversion of proteins to the prion state is protein dependent. It has been shown that prions extracted from cells can serve as "seeds" for prion formation in vitro.
One of the most well-studied proteins prone to prion formation in yeast is translation termination factor (eRF3), which forms so-called PSI cells. Such cells have an altered physiological state and altered levels of expression of certain genes, which has led to the hypothesis that prion formation in yeast may play an adaptive role [1] .
Diagnostics
Diagnosis of prion diseases is based on the detection of cognitive disorders, their progressive development, and already at an intermediate stage of development - non-specific EEG disturbances, gradually turning into specific patterns (burst suppression pattern) with myoclonus in the clinical and EMG picture, and epileptic seizures.
Only 14-3-3 protein is present in the cerebrospinal fluid.
In sporadic Creutzfeldt-Jacobs disease, T2-weighted MRI demonstrates hyperintensity in the nucleus basalis and caudalis. In the new variant of the disease, it is present in the pulvinar thalamus and has the shape of a club.
DNA testing can help identify genetic and familial forms of the disorder, which means identifying the E200K mutation.
The clinical diagnostic conclusion is confirmed by immunohistochemical and neurohistological examination of brain tissue.
- typical neurocytic vacuolization;
- spongiomatous degeneration of the neuropil;
- loss of nerve cells;
- astrogliosis;
- presence of scrapie-associated fibrils in the affected brain.
Etiology
A person can become infected with prions contained in food, since they are not destroyed by enzymes in the digestive tract. Freely penetrating through the wall of the small intestine, they ultimately enter the central nervous system. This is how a new variant of Creutzfeldt-Jakob disease (nvCJD) is transmitted, which people become infected with after eating beef containing nerve tissue from cattle with bovine spongiform encephalopathy (BSE, mad cow disease).
Prions can also enter the body parenterally. Cases of infection have been described through intramuscular administration of drugs made from human pituitary glands (mainly growth hormones for the treatment of dwarfism), as well as infection of the brain with instruments during neurosurgical operations, since prions are resistant to currently used thermal and chemical methods of sterilization. This form of Creutzfeldt-Jakob disease is designated iatrogenic (1CJD).
Under certain, unknown conditions, spontaneous transformation of the prion protein into a prion can occur in the human body. This is how the so-called sporadic Creutzfeldt-Jakob disease (sCJD) arises, first described in 1920 independently by Hans Gerhard Creutzfeldt and Alphonse Maria Jakob.
It is believed that the spontaneous occurrence of this disease is due to the fact that normally a small number of prions are constantly generated in the human body, which are effectively eliminated by the cellular Golgi apparatus. Violation of this ability of “self-cleaning” of cells can lead to an increase in the level of prions above the permissible normal limit and to their further uncontrolled spread. According to this theory, the cause of sporadic Creutzfeldt-Jakob disease is a dysfunction of the Golgi apparatus in cells.
A special group of prion diseases are hereditary (congenital) diseases caused by a mutation in the prion protein gene, which makes the resulting prion protein more susceptible to spontaneous changes in spatial configuration and their transformation into prions. This group of hereditary diseases also includes the hereditary form of Creutzfeldt-Jakob disease (fCJD), which is observed in a number of countries around the world.
In prion pathology, the highest concentration of prions is found in the nervous tissue of infected people. A significant number of prions are found in lymphatic tissue. The presence of prions in biological fluids, including saliva, has not yet been unequivocally confirmed. If the idea of a constant occurrence of small numbers of prions is correct, then it can be assumed that new, more sensitive diagnostic methods will discover these numbers of prions scattered throughout various tissues. In this case, however, we will talk about the “physiological” level of prions, which do not pose any threat to humans.
How to slow down the progression of prion disease
Until recently, it was difficult to slow down prion disease, let alone cure it.
Modern neurosurgery can alleviate degenerative complications by transplanting neurons for prion disease.
A revolutionary attempt to stop pathogenic proteins was pioneered by a team of doctors from the University of Pittsburgh. Dr. Kondziolka's team transplanted neurons in vitro (2-6 million LBS neurons) into 12 patients as part of the study. 12 months after surgery, none of the recipients had any adverse effects from the transplant, and all received new cells. In 6 patients there was a significant improvement in condition.
Several years ago, the journal Nature reported on the development of antibodies against prions in England by a team of scientists led by Simone Hawke (Imperial College). In tests on infected mice, rodents that received the antibodies were cured within a month of infection.
Doctors intend to use this opportunity to clear neurons of pathogenic proteins to treat a new variant of Creutzfeldt-Jacobs disease in humans. Further research should help improve drugs that can clear prions from the brain and its microglia.
Recently, M. Pfeiffer and several other German scientists discovered and confirmed a method to clear the brain of prions. They proved that RNA interference can successfully treat diseases by removing from the RNA cell the part responsible for the production of pathogenic proteins. This prevented them from mutating into dangerous proteins.
A similar method is the effect of artificially synthesized primers (oligonucleotides) against prions. Like RNA interference, primer design is under investigation.
Theoretically, the PrP13 peptide, similar to the structural product found in these diseases, may be promising in the treatment of diseases. The substance targets the PRNP gene, which should affect the conversion of PrPC to PrPres. The last pathogenic molecule named changes to a PrPC-like structure. The substance goes by the acronym PrP13, and animal studies have been able to prolong survival by 50-300%.
What are prions and what is their mechanism of action on the body (modern ideas)?
In fact, humans and many other living things contain PrPC proteins. In Russian - the normal form of prion proteins (they were discovered after Siggurdson’s research, which is why the name is so strange). Its length, amino acid sequence, and secondary structure are known. It is important to know that the final structure consists of three α-helices and a double-stranded antiparallel β-sheet.
They have an interesting property, namely they are precipitated by high-speed centrifugation, which is a standard test for the presence of prions. There is evidence that PrP plays an important role in cell attachment and the transmission of intracellular signals, and therefore may be involved in the communication of brain cells. However, the functions of PrP are not well understood.
(a) normal (b) pathology
Experiments in mice lacking these proteins show that the absence of PrP leads to nerve demyelination. It is possible that prion proteins normally support long-term memory.
But this is normal.
Sometimes “problems” happen and proteins called PrPSc appear - infectious prions. They differ in that instead of α-helices, β-sheets predominate in them.
This leads to a change in the interaction of other proteins with the new protein. It wouldn’t be so bad if only one protein was formed per organism. The trouble is that, once formed, the protein (!) itself begins to change the structure of others. Let's consider the basic mechanisms of PrPSc reproduction
It is believed that prion disease can be acquired in 3 ways: through direct infection, hereditary or sporadic (spontaneous) or combinations thereof.
Sporadic (that is, spontaneous) prion disease occurs in a population in a random individual. This is, for example, the classic version of Creutzfeldt-Jakob disease. There are two main hypotheses regarding the spontaneous occurrence of prion diseases. According to the first of them, a spontaneous change occurs in the hitherto normal protein in the brain, that is, a post-translational modification takes place.
An alternative hypothesis is that one or more cells in the body at some point undergo a somatic mutation (that is, not inherited) and begin to produce the defective PrPSc protein. Be that as it may, the specific mechanism for the spontaneous occurrence of prion diseases is unknown.
The second is infection. According to modern research, the main way to acquire prion diseases is by eating contaminated food. It is believed that prions can remain in the environment in the remains of dead animals, and are also present in urine, saliva and other body fluids and tissues (blood, cerebrospinal fluid).
Because of this, infection with prions can also occur during the use of unsterile surgical instruments. This makes it difficult to sterilize surgical instruments or devices in the slaughterhouse. Prions are generally resistant to proteases, heat, radiation, and storage in formalin, although these measures reduce their ability to become infected.
Effective disinfection against prions must involve hydrolysis or damage/destruction of their tertiary structure. This can be achieved by treating with bleach, sodium hydroxide and strongly acidic detergents. Spending 18 minutes at 134°C in a sealed steam autoclave will not deactivate prions.
Ozone sterilization is currently being studied as the main modern method for deactivating and denaturing prions. Renaturation of a completely denatured prion to an infectious state has not been recorded, but for partially denatured prions under some artificial conditions this is possible.
It is also worth remembering that these proteins can remain in the soil for a long time due to binding to clay and other soil minerals. Don't get paranoid, but theoretically they could be everywhere.
https://www.youtube.com/watch?v=userprightcom
In 2011, the discovery of prions transmitted through the air in aerosol particles (i.e., airborne droplets) was reported. Also in 2011, preliminary evidence was published that prions can be transmitted by urine-derived human menopausal gonadotropin, used to treat infertility.
Theoretically, with the help of just one sick animal with prion disease, it is possible to destroy entire nations and countries by simply adding its bone meal to feed additives and selling them to the desired state. A similar situation occurred in the late 80s in Britain (the mad cow disease epidemic). Then, most likely out of ignorance (and not out of malicious intent), the above process occurred, which claimed the lives of about 200 people (as of 2009) and 179 thousand heads of cattle.
Prion propagation
The third mechanism is genetic. It was opened recently and does not fit into the overall picture at all. The gene encoding the normal PrP protein, PRNP, localized on chromosome 20, was identified. In all hereditary prion diseases, a mutation of this gene occurs.
A “distorted” prion protein that has entered in one way or another begins to change the structures of proteins close to it in structure, turning them into the same pathogenic agents.
The main hypothesis that most closely reflects this process is very simple. One molecule of PrPSc attaches to one molecule of PrPC and catalyzes its transition to the prion form. The two PrPSc molecules then separate and continue to convert other PrPCs into PrPSc.
But the scheme raises more questions than answers.