Прионные болезни – трансмиссивные спонгиоформные энцефалопатии (ТСЭ) – это относительно новая группа дегенеративных заболеваний ЦНС. Расстройства характеризуются хроническим необычным взаимодействием иммунной системы и предполагаемого этиологического фактора – белка приона.
Он на периферическом пути болезни размножается в эндоретулярной системе, откуда переходит в ЦНС (головной и спинной мозг), где он вызывают васкуляризацию нейроцитов, глиальных клеток, спонгиоматозную реконструкцию нейропилуса, вымирание и уменьшение количества нейрональных клеток, глиоз без значительного вовлечения воспаления.
Прион — убийца нейрона
Прион, также известный в микробиологии как инфекционный PrP Sc , – это профессиональный термин, описывающий дефектную форму протеина, часто встречающегося в тканях мозга млекопитающих, способствующего долгосрочному функционированию памяти.
Прионы убивают нейроны очень быстро, что провоцирует стремительное развитие дегенеративных заболеваний.
Согласно теории, речь идет об источнике многих нарушений ЦНС животных (включая человека). Эти белки играют роль в наследственных изменениях и эволюции.
Один из регуляторных факторов прионопреобразования – шапероны, функция которых заключается в чистке клеток от патогенных белков. Как шапероны, так и прионы относятся к группе клеточных протеинов.
Теории и гипотезы
Теория, объясняющая, почему прионы убивают нейроны, была сформулирована профессором С. Б. Прусинером в 1982 г. Он также впервые использовал слово «прион» (первоначально предназначалось использование обозначения «проин», как сочетания слов «proteinaceous» и «infectious», но в измененном состоянии оно звучало лучше).
Теорию прионов, как патогенных белков, Прусинер сформулировал в связи с поиском возбудителя расстройства Крейтцфельда-Якоба. Обнаруженный ним агент являлся чем-то новым. До 1982 г. считалось, что инфекционные заболевания могут вызывать только инфекционные организмы, содержащие нуклеиновую кислоту, несущую генетическую информацию. Но строение белка не содержит нуклеиновой кислоты. Это белок, размножающийся, изменяя аналогичные белки в организме. В 1997 г. С. Б. Прусинер получил Нобелевскую премию в этой области.
Патогенные белки прионы имеют одинаковую первичную структуру (аминокислотную последовательность), но различаются по своей конформационной конфигурации. В то время как дикий PrP с имеет выраженное преобладание α-геликса и около 5% β-листа, патогенный PrP Sc (Sc – scrapie) имеет долю β-листа до 40%. Причины аберрантных свойств прионов PrP Sc все еще рассматриваются. Сегодня есть несколько гипотез.
- Вирусная гипотеза объясняет воздействие прионов вирусологией. Предполагается участие РНК-вирусов в трансмиссивных губчатых энцефалопатиях. Как вироиды, так и прионы – мелкие инфекционные патогены; следовательно, результат воздействия вируса – прион инфекционной природы.
- Мультикомпонентная гипотеза. Предполагается, что для образования инфекционных белков, как новых возбудителей болезней необходима связь с полианионами и липидами.
- Отравление тяжелыми металлами. Интоксикация вызывает развитие инфекции при недостатке или избытке в организме меди (для здорового протеина необходимо оптимальное количество меди).
В чем заключается токсичность белка для нейрона?
После появления инфекционного протеина он может «запечатлеть» свою конформацию в здоровые нейроны. Токсические протеины могут убивать митохондрии в нейронах, распространяя болезнь на ткани.
Патогенные белки чрезвычайно устойчивы к физическим и химическим воздействиям, что приводит к трудностям при стерилизации (были предприняты попытки сжигания головного мозга у пораженных животных при 600°C, пеплом впоследствии были заражены примерно 1/3 животных). В соответствии с тем, какие метаболические процессы нарушают прионы в нейроне, наиболее инфекционные – ткани глаза, головного и спинного мозга.
Scisne?
Молекулярный биолог Байрон Кои о структуре прионов, прионных болезнях, их причинах и лечении.
Болезнь Крейтцфельдта — Якоба |
Прионы составляют отдельный класс инфекционных агентов; они имеют белковую основу и не содержат генома, состоящего из нуклеиновых кислот. Концепция существования прионов включает в себя идею о новой, белковой наследственности — измененных формах белков в организме-хозяине, которые при переносе в новый организм могут вызвать в получателе изменение фенотипа. Большинство прионов являются патогенами, но по сравнению с другими классами патогенов (например, вирусами, бактериями, грибами, паразитами) они уникальны тем, что распространяются внутри и между хозяевами без переноса или репликации собственных ДНК или РНК.
Прионы, как правило, — это повторно свернутые и агрегированные белки, которые распространяются, внедряясь в организм-хозяина и стимулируя в нем рефолдинг соответствующей нормальной формы белка. Прионный агрегат растет, а затем каким-то образом фрагментируется, чтобы сгенерировать больше прионных агрегатов. Во многих отношениях это похоже на лед-девять Курта Воннегута: рост прионов аналогичен росту кристаллов, а сами прионы часто описывают как затравку по аналогии с затравочными кристаллами. Таким образом, прионам не нужно переносить свой генетический код, но организм-хозяин должен создать нормальный белок, из которого состоят патогенные прионы. Многие белки (если не большинство) могут перегруппироваться и/или собираться в упорядоченные агрегаты, которые в определенных естественных или экспериментальных условиях — в тканях или пробирке — могут расти, однако не все такие белковые агрегаты являются патогенными прионами. Термин «прион» означает, что на биологическом уровне структура повторно свернутого белка может распространяться между хозяевами или по меньшей мере из клетки в клетку внутри многоклеточного организма-хозяина. Недавно звучало предложение собрать под зонтичным термином «прионы» все состояния белка, которые способствуют его росту в форме мультимерных скоплений, но это расширенное использование термина нарушает лежащую в его основе идею передаваемости и не позволяет выделить прионы из многих других клеточных структур, которые могут расти, но не имеют тенденции распространяться на другие клетки и организмы.
История исследований
Прототипом прионной болезни было трансмиссивное нейродегенеративное заболевание овец — таинственная смертоносная скрейпи (или почесуха овец). Ранние исследования показали, что агент скрейпи необычайно устойчив к лечению, которое нейтрализует другие патогены, и может годами оставаться на пастбищах. То, что агент скрейпи проявляет устойчивость, в частности, к радиации, привело к тому, что в 1960-х годах Дж. С. Гриффит и Тиквах Альпер предположили, что он представляет собой новый класс патогенов, который не имеет собственного нуклеинового генома и может быть аномальной самовоспроизводящейся формой белка или мембраны. Между тем описания патологии мозга, вызванной человеческой болезнью куру в Папуа — Новой Гвинее, которые выполнил Карлтон Гайдушек, привели Уильяма Хэдлоу к мысли, что куру похожа на скрейпи овец, и Хэдлоу порекомендовал, чтобы куру испытали на передаваемость от людей к другим приматам. Гайдушек успешно проделал эту работу и показал, что люди племени форе заболевали куру во время ритуальных каннибалистских праздников. Яркой особенностью куру и других прионных заболеваний, часто скрывавшей их причины, является длительный инкубационный период между заражением и появлением клинических признаков, который у людей может превышать четыре десятилетия.
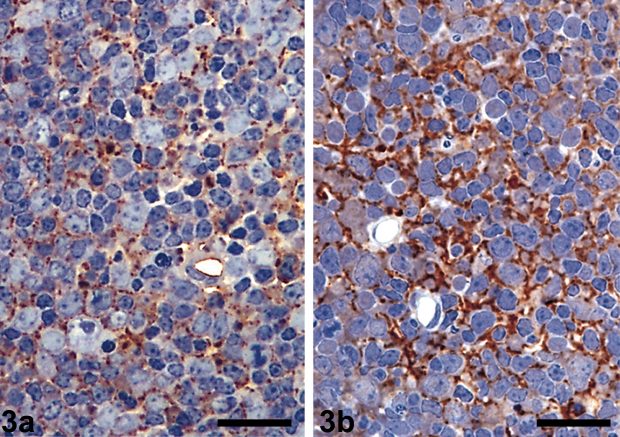
Лимфатические узлы здоровой (а) и инфицированной (b) овцы — окрашивание при помощи тел четко демонстрирует признаки прионов болезни скрейпи в тканях инфицированной овцы / wikipedia.org |
В 1980-х годах Стэнли Прузинер придумал для этих агентов термин «прион» и первым определил специфический белок, который является основным компонентом прионов скрейпи, — прионный белок (PrP). Гомологи того же белка были обнаружены у многих видов млекопитающих, в том числе и у людей, а аномальные агрегаты PrP выявлены в других трансмиссивных нейродегенеративных заболеваниях людей и животных, подобных скрейпи. Эти заболевания теперь известны как прионные заболевания, или трансмиссивные губчатые энцефалопатии. Стэнли Прузинер, Чарльз Вайсман и другие исследователи показали, что PrP является важным фактором восприимчивости прионных заболеваний.
Моя лаборатория в сотрудничестве с Питером Лэнсбери показала, что связанные с заболеваниями формы PrP сами могут вызвать трансформацию нормальных молекул PrP в аномальные формы. В этих реакциях превращения мы выявили поразительные биохимические особенности, которые помогли объяснить характеристики известных штаммов прионов и барьеры их передачи между разными видами. Однако, чтобы однозначно доказать, что прионы состоят из повторно свернутых агрегатов PrP и им не нужны специфически-прионные нуклеиновые кислоты, потребовалось разработать методы непрерывной бесклеточной амплификации прионов или реакции образования прионов de novo. Они первоначально были созданы лабораториями Сото, Супаттапоне и Прузинера в 2000-х годах; до того времени было трудно полностью исключить вероятность того, что эти заболевания вызваны неопознанными вирусами.
Хотя слово «прион» впервые было применено к описанным выше трансмиссивным губчатым энцефалопатиям, первые однозначные доказательства того, что в биологии существуют инфекционные белки, были получены, когда Рид Уикнер в 1994 году понял, что некоторые необъяснимые эпигенетические элементы в дрожжах — это прионы. Эти прионы состояли не из гомолога PrP, а из совершенно особенных белков дрожжей. Относительная простота и мощь биологии и генетики дрожжей позволили Уикнеру и другим исследователям ясно продемонстрировать ряд основополагающих принципов прионной биологии и структуры, которые было гораздо сложнее выявить на прионах млекопитающих.
Методы исследования
К сожалению, многие из стандартных методов, на которых долгое время базировались исследования обычных патогенов, — генетика патогенов, серология, рентгеноструктурный анализ, спектроскопия ядерного магнитного резонанса (ЯМР) — чрезвычайно трудно применить к прионам. Без каких-либо специфических патогенных генов, которые можно было бы секвенировать или подвергнуть мутации, многие стандартные генетические и обратные генетические подходы к выявлению структуры и функции патогенов не работают. Поскольку прионы состоят из белков организма-хозяина, иммунный ответ хозяина на патоген очень мал; таким образом, провести простое серологическое обнаружение прионных инфекций, основанное на взаимодействии с антителами, очень сложно. Кроме того, прионы млекопитающих, как правило, плотно упакованы, сильно гликозилированы и связаны с другими молекулами организма-хозяина, и поэтому даже специфические прионные конформационные эпитопы (поверхности, распознаваемые антителами) на агрегатах PrP трудно обнаружить и использовать. Все попытки определить трехмерные структуры прионов на протяжении уже долгого времени заходят в тупик, так как очищенные прионы имеют агрегированный, но некристаллический характер.
В течение многих лет единственным способом обнаружения и анализа прионов млекопитающих был биоанализ животных, который даже на самых быстрых моделях — грызунах — длился от нескольких месяцев до одного года. В конкретном организме разные штаммы обычно можно различить по периодам инкубации, невропатологическим паттернам и биохимическим признакам связанных с болезнью отложений PrP или прионов.
К счастью, в последнее время были разработаны мощные бесклеточные амплификационные анализы прионов, такие как циклическая амплификация прионной формы белка (PMCA), вибрационно-индуцированный конверсионный анализ в режиме реального времени (RT-QuIC) и анализ клеток скрейпи. Эти методы основаны на присущем прионам механизме репликации. И PMCA, и RT-QuIC чрезвычайно чувствительны: они могут усилить присутствие прионов в триллион раз, почти до точки обнаружения нескольких прионных частиц. Реакции PMCA распространяют инфекцию прионов, тем самым отражая и освещая многие аспекты прионной биологии, в то время как анализы RT-QuIC, как правило, не распространяют полностью инфекционные прионы, но обеспечивают более быстрые, более практичные и более высокопроизводительные методы их обнаружения, и, таким образом, они стали самыми современными инструментами в диагностике прионных заболеваний. Как PMCA, так и RT-QuIC в некоторых случаях помогают различать важные штаммы прионов у определенных видов организмов-хозяев.
В выявлении базовой структуры прионов наблюдается медленный прогресс. При помощи полупроводниковых ЯМР-исследований была обнаружена молекулярная архитектура некоторых прионов грибов и прионоподобных фибриллярных структур PrP млекопитающих. Электронная кристаллография, дифракция волокон и криоэлектронные микроскопические исследования помогли описать ключевые структурные ограничения прионов млекопитающих, но применение этих и, возможно, других структурных биологических методов еще нужно улучшить.
Структура и воспроизведение прионов
Разобраться в структуре и механизмах репликации прионов млекопитающих, по крайней мере на молекулярном уровне, крайне сложно. Сначала нужно объяснить, как неправильно свернутые белки могут распространяться в роли патогенов, не перенося своего собственного нуклеинового генома. Затем следует также объяснить, как белки с единой последовательностью аминокислот, такие как PrP того или иного животного-хозяина, могут образовывать разные штаммы прионов, которые исправно распространяются и вызывают различные фенотипы болезни без генетических мутаций, объясняющих вариации штаммов в обычных патогенах.
Множество исследований указывает на то, что прионы млекопитающих — это упорядоченные скопления нескольких молекул PrP, плотно упакованных и часто фибриллярных или нитевидных. Молекулы PrP (мономеры) в прионах по сравнению с нормальными свободными молекулами PrP пересвернуты практически полностью. Когда правильные молекулы PrP включаются в растущие прионные агрегаты, эти агрегаты вызывают их рефолдинг, причем прионы действуют как штамм-специфические шаблоны или затравки, которые каким-то образом придают свои собственные аберрантные формы каждой входящей молекуле, контролируя стабильную репликацию своего штамма.
За рамками этого грубого описания детали структуры и распространения прионов на молекулярном уровне остаются неясными. Также нерешенным остается вопрос о том, как прионы распространяются за пределы исходного места заражения в организме-хозяине. Существующие данные свидетельствуют о том, что наиболее эффективная межклеточная передача прионов связана с мембранозными структурами, такими как экзосомы или туннелирующие нанотрубки, — скорее всего, потому, что прионы обычно связаны с мембранами липидными якорями; однако возможность этих мембранных структур способствовать распространению прионов in vivo еще предстоит определить. Очень важно понять механизмы распространения прионов, поскольку способности различных неправильно свернутых белковых агрегатов распространяться внутри и между клетками, тканями и индивидами определяют то, действуют ли они как инфекционные патогены или являются относительно безобидными сбоями белкового метаболизма.
Прионные болезни
Многие виды млекопитающих, включая людей, низших приматов, крупный рогатый скот, овец, коз, оленей, лосей, кошек, норок, грызунов и различных экзотических копытных, восприимчивы к прионным заболеваниям PrP. Но такими являются не все виды: собаки и лошади, судя по всему, представляют собой заметные исключения. Разные виды обычно экспрессируют несколько разные нормальные молекулы PrP, и различия в аминокислотной последовательности PrP могут сильно влиять на восприимчивость хозяина к входящим прионным инфекциям. Например, люди, как известно, до некоторой степени восприимчивы к губчатой энцефалопатии крупного рогатого скота (ГЭКРС), но, по-видимому, устойчивы к скрейпи овец и, насколько нам известно, хронической изнуряющей болезни оленей. По какой-то причине лесные полевки и беличьи обезьяны необычайно восприимчивы к широкому спектру прионных инфекций других видов.
Механизмы, с помощью которых прионные инфекции вызывают нейродегенеративные заболевания, нам пока неизвестны. Агрегаты различных прионных штаммов в организмах-хозяевах разных видов могут накапливаться преимущественно в разных областях центральной нервной системы и вызывают ряд невропатологических расстройств. Очевидно, что конечным эффектом по крайней мере частичного повреждения является сбой в работе нейронов и их потеря, что вызывает множество клинических симптомов и приводит к летальному исходу. Известно, что ряд нейрофизиологических процессов и путей нарушается, но многое еще предстоит определить относительно того, связаны ли такие нарушения с прямой или косвенной токсичностью прионов и в какой степени та или иная недостаточность или комбинация недостаточностей наиболее ответственна за кончину больного.
У людей причины прионных заболеваний могут быть генетическими (из-за специфических мутаций гена PrP), приобретенными (вызванными заражением — например, воздействием куру, ГЭКРС или другим содержащим прионы материалом) или спорадическими (неизвестного происхождения; обычно предполагается, что они обусловлены спонтанным образованием прионов у конкретного индивидуума). Подавляющее большинство прионных заболеваний человека являются спорадическими, и среди них наиболее распространена спорадическая болезнь Крейтцфельдта — Якоба (sCJD), заболеваемость которой в год во всем мире составляет примерно один случай на миллион населения. Ряд различных мутаций в гене PrP может вызывать множество семейных прионных заболеваний, при этом некоторые мутации являются полностью пенетрантными (всегда вызывающими болезнь у носителей мутации), а другие — менее пенетрантными. Клинические симптомы и прогрессирование болезни могут заметно различаться в разных организмах-хозяевах и при разных типах прионных заболеваний, но могут включать деменцию, расстройство координации, бессонницу, галлюцинации, жесткость мышц, спутанность сознания, усталость и затрудненность речи.
Существуют также важные прионные заболевания животных. ГЭКРС возникла как масштабная эпидемия крупного рогатого скота в связи с тем, что можно было бы назвать «сельскохозяйственным каннибализмом» в 1990-х годах. Потребление ГЭКРС — зараженной говядины — вызвало тогда почти двести случаев варианта болезни Крейтцфельдта — Якоба у людей, однако превентивные меры почти ликвидировали ГЭКРС и предотвратили появление новых случаев. Хроническая изнуряющая болезнь оленей прокатывается по Северной Америке с угрожающей скоростью, причем случаи болезни также возникают в Южной Корее и Норвегии. Скрейпи — это постоянная проблема с овцами и козами во многих частях мира.
Диагностика и лечение прионных болезней
В последнее время были достигнуты значительные успехи в том, чтобы точно и относительно неинвазивно диагностировать прионные заболевания человека у живых пациентов на основе новых прион-специфических тестов мазков из носа, спинномозговой жидкости, крови, мочи или кожи. Например, RT-QuIC-тестирование спинномозговой жидкости и/или материалов назальной щеточной биопсии может достигать 100% точности при диагностике спорадической болезни Крейтцфельдта — Якоба. Эти тесты выгодны потому, что измеряют возбудителей прионной болезни, но они еще не полностью проверены и не рекомендованы официально такими организациями, как ВОЗ. В остальном диагностика спорадических прионных заболеваний у людей зависит в первую очередь от совокупности клинических признаков, результатов сканирования мозга, электроэнцефалограмм и других биомаркеров, которые вместе могут иметь высокую диагностическую чувствительность, но не полностью специфичны для прионных болезней.
Несмотря на описанные выше недавние успехи в разработке новых прионных тестов, действующие руководящие принципы таковы: для окончательной диагностики спорадического или приобретенного прионного заболевания необходимо невропатологическое исследование тканей головного мозга, полученных в результате биопсии (что редко) или аутопсии. Полагаю, в скором времени эти рекомендации будут изменены, в них будут включены новые, менее инвазивные прижизненные тесты для выявления прионов. К сожалению, несмотря на то, что этот прогресс в раннем диагностировании прионных болезней должен улучшить перспективы разработки и применения терапевтических средств, в настоящее время доступных методов лечения, которые доказали бы свою эффективность в клинических испытаниях, не существует.
Открытые вопросы и будущие направления исследований
В области прионных заболеваний млекопитающих остаются открытыми следующие ключевые вопросы: «Какова самораспространяющаяся структура прионов и как она варьируется в зависимости от штамма приона?», «Как деятельность прионов приводит к повреждениям мозга?», «Как мы можем предотвратить эти повреждения или восстановить их при лечении прионных заболеваний?», «Каковы наиболее актуальные механизмы передачи прионных заболеваний у людей и животных?», «Какие прионные заболевания животных (помимо ГЭКРС), если такие существуют, имеют зоонозный потенциал, то есть могут вызывать болезнь у людей?», «До какой степени другие патогенные, неправильно свернутые белки, которые также могут выступать в качестве затравки, ведут себя как основанные на PrP прионы в своей способности распространяться внутри или между людьми, вызывая болезнь?»
Действительно, последний вопрос представляет собой важный рубеж в изучении многих заболеваний, связанных с неправильным образованием белков, особенно тех, которые связаны с патогенным накоплением аномальных фибриллярных белковых отложений (например, амилоидных фибрилл и бляшек). Эти болезни включают в себя болезни Альцгеймера, Паркинсона и Гентингтона, а также боковой амиотрофический склероз, лобно-височные деменции, хроническую травматическую энцефалопатию и диабет второго типа. Различные белки организма-хозяина образуют скопления при этих и многих других заболеваниях, но, как и прионы, такие скопления обычно растут за счет включения в затравку нормальных растворимых молекул белка. Таким образом, потенциал для прионоподобного распространения белков существует на молекулярном уровне. Также растет количество доказательств того, что множество различных связанных с болезнями белковых отложений может расти и распространяться так же, как и прионы, вызывая патологии после инокуляции в локализованные участки у подопытных животных.
Результаты этих исследований поднимают насущные вопросы о том, могут ли многочисленные заболевания, основанные на повторном фолдинге белка, — а они зачастую гораздо более распространены, чем прионные заболевания, основанные на PrP, — быть переданы людям или животным в реальных условиях. Болезнь Крейтцфельдта — Якоба передается между людьми через трансплантацию тканей, инъекции гормонов, полученных от трупов, переливание крови и зараженные медицинские инструменты. Вторым фактором в таких ятрогенных передачах является тот факт, что прионы часто не полностью инактивируются стандартными процедурами клинической дезинфекции.
Еще предстоит установить, могут ли быть определены другие типы потенциально прионоподобных, ассоциированных с болезнями агрегатов белка, которые также могут быть устойчивы к инактивации и при этом способны инициировать или ускорять патогенные процессы у людей. Я не знаю никаких эпидемиологических указаний на то, что это так, но дальнейшее тщательное изучение этого вопроса кажется оправданным.
Байрон Кои — Ph.D., Chief of the TSE/Prion Biochemistry Section of the Laboratory of Persistent Viral Diseases, National Institute of Allergy and Infectious Diseases. ПостНаука
Факторы риска развития инфекций
Фактор риска заболеваний с генетической точки зрения – наличие определенной последовательности оснований в определенном месте гена, кодирующего белок.
Высокий риск формирования инфекционных прионов и прионных болезней представляют операции с загрязнением тканей и трансплантацией от пациента с присутствующей инфекцией.
Недоказанный риск – потребление тканей больного животного. Здесь важен фактор токсичности приона, его свойства выживать при воздействии температуры, в несколько раз превышающей температуру кипения воды. Животноводство без потребления продуктов не является рисковым.
Наиболее опасные (заразные) из пораженного организма – мозг (ЦНС), мозговые оболочки, роговица (глаз), кровь, лимфатические органы. Мышцы и другие органы представляет намного меньший риск, но не исключают его.
Пути заражения
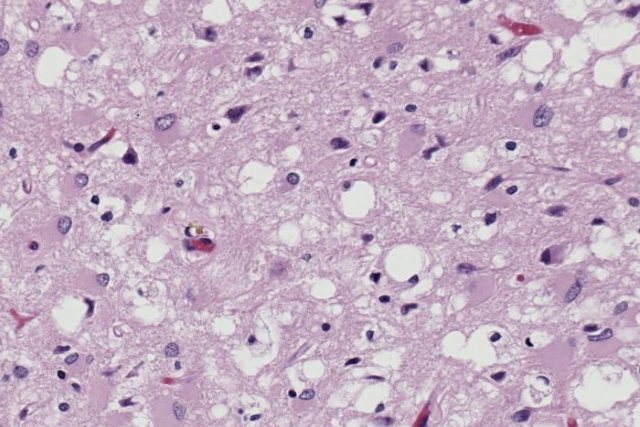
Гистологический препарат, поражение ткани прионами с образованием характерной губчатой структуры.
https://www.youtube.com/watch?v=upload
Очень мало известно о молекулярном характере прионов, вызывающих заболевания. Заражение могут вызвать примерно 100 000 молекул, которые в большинстве случаев образуют большие скопления. Значение агрегации отдельных молекул в ассоциации для вирулентности прионов пока не известна. Нельзя исключить, что вирулентными являются и отдельные молекулы прионов.
Из некоторых экспериментов следует, что для возникновения прионов в ткани достаточно лишь временного контакта ткани с материалом, содержащим прионы, и нет необходимости, чтобы прионы были навсегда внесены в организм. Этот риск является актуальным, например, в связи с использованием хирургических инструментов, заражённых прионами.
Ход болезни и распространение прионов по организму зависит от типа приона. Прионы отличаются составом аминокислот, характерных для данного вида, определяемых видовым геном прионового протеина, а также так называемыми посттрансляционными модификациями или степенью гликозилирования базовой белковой цепочки.
Посттрансляционная модификация значительно влияет на характеристики прионов и именно ей приписывают разницу между так называемыми прионовыми родами. В случае нового варианта (nvCJD) был пока что описан лишь один вид приона, сходный с прионами скота, заражённого бычьей губчатой энцефалопатией. Поэтому течение заболевания у человека и животных, заражённых новым вариантам, практически одинаково.
У прочих видов живых существ, однако, известно много прионовых родов. У овец были описано примерно два десятка таких родов, которые не вирулентны для человека. Течение овечьего прионового заболевания в зависимости от рода прионов и драматически отличается — от очень быстрого, с практически внезапной гибелью, до медленного, затяжного.
Нетипичные случаи клинического течения нового варианта у скота, заражённого бычьей губчатой энцефалопатией, которые имели место в Японии и Италии, наводят на мысль о существовании большего количества родов бычьих прионов. Если бы этот род бычьих прионов попал в организм человека, следовало бы ожидать возникновение нового варианта с симптомами и клиническим течением, отличными от известных случаев.
У пациентов, больных болезнью Крейтцфельдта-Якоба, прионы распространяются в нервной системе, тканях глаза и лимфатических тканям, включая миндалины, селезенку, а также в слепой кишке. Наибольшее количество прионов находится в нервной системе, а наименьшее — в лимфатической ткани.
Пока что не был зарегистрирован ни один случай переноса нового варианта болезни Крейтцфельдта-Якоба (nvCJD) при медицинском вмешательстве, что является, разумеется, хорошей новостью. С другой стороны, специалисты предупреждают о преувеличенном оптимизме, прежде всего в условиях Великобритании, так как инкубационный период может быть достаточно долгим (от 5-8 месяцев до 10-15 лет).
Характеристика инфекций
Заболевания вызываются возбудителями, не содержащими нуклеиновой кислоты. Они могут быть спорадическими, генетически обусловленными, инфекционными (даже ятрогенными). Присутствие дефектных белков вызывает расстройства, обычно называемые спонгиоформными энцефалопатиями. Это дегенеративные нарушения нервной системы, при которых мозг постепенно приобретает губчатый вид из-за миниатюрных отверстий, ввиду того, что прионы убивают нейроны.
Все инфекции неизлечимы и смертельны. Сегодня проводятся исследования и тестирования, направленные на изучение методов замедления течения прионной болезни.
Прионы и медицинские инструменты
Прионы очень стойки к обычным методам дезинфекции. Ионизирующее, ультрафиолетовое или микроволновое излучение на них практически не действует. Дезинфекционные средства, обычно используемые в медицинской практике, действуют на них лишь в очень ограниченной мере. Надёжно их ликвидируют дезинфицирующие реактивы — сильные окислители, разрушающе действующие на протеины.
Другое затруднение представляет собой стойкость прионов к высоким температурам. Даже при автоклавировании при 134 °C в течение 18 минут невозможно достичь полного разрушения прионов, и прионы «выживают» в форме, способной вызвать заражение. Стойкость к высоким температурам ещё более возрастает, если прионы засохнут на поверхности металла или стекла или если образцы перед автоклавированием были подвергнуты действию формальдегида.
В Великобритании, где новый вариант является очень серьёзной проблемой, по этим причинам уже используются одноразовые хирургические инструменты для тонзиллэктомии. В будущем напрашивается альтернативное решение: создания новых инструментов, с учётом повышенных требований к очистке и обеззараживанию.
Намного более сложным решением этой проблемы является лечение пациентов группы риска. К ним относятся пациенты, которые подверглись операциям, при которых была использована потенциально заражённая твёрдая мозговая оболочка, или пациенты из семей с наследственной формой болезни Крейтцфельдта-Якоба. ВОЗ в этом случае не требует никаких специальных мер.
Отдельные нозологические единицы
Прионные болезни человека видоспецифичные. В основном они распространяются только среди особей одного вида. Но учеными была продемонстрирована межвидовая передача (преодоление межвидового барьера): это произошло у скрепи овец с ее передачей коровам (коровье бешенство). Передача заболевания от животного к человеку, несмотря на многие подозрения, не была продемонстрирована.
Ряд заболеваний имеет наследственное происхождение, часть является инфекционной. Иногда болезнь возникает неожиданно, без возможности профилактики.
Болезнь существовала только в Папуа-Новой Гвинее в племенах людоедов, потреблявших мозги мертвых. Она проявляется тремором («куру» означает «тремор»), нарушениями речи, равновесия (атаксия). Через несколько месяцев наступает смерть. После остановки каннибализма, заболевание куру исчезло.
Болезнь Крейцфельдта-Якоба (Creutzfeldt-Jakob disease)
Это редкое расстройство, поражающее пожилых людей (чаще всего 50-70-летних). Его течение относительно быстрое, смерть наступает через несколько месяцев (до года) от проявления первых симптомов. Нейроны быстро гибнут, развивается прогрессивная деменция, не имеющая известного лечения. Существует некоторая наследственная предрасположенность к этому заболеванию:
- Спорадическая форма. Заболеваемость составляет 1-2/1000000. Проблемы начинаются примерно в возрасте 65 лет. Расстройство возникает в виде острого прогрессирования деменции (в течение 2-3 месяцев), атаксии, миоклонуса. Возможностей, как продлить жизнь нейронам при этом прионом заболевании, нет, пациент умирает через 5-12 месяцев после появления первых симптомов.
- Ятрогенная форма. Возникала у пациентов, получавших гормон роста человека из трупных гипофизов (сегодня он готовится рекомбинантно), пересадкой твердых оболочек, перикарда, роговицы. Существует риск нейрохирургической передачи. Заражение возможно также при переливании.
- Семейная форма. Это генетическая форма с мутацией в гене PRNP и психоневрологической симптоматикой.
- Новый вариант. Характеризуется психиатрическими симптомами (тревога, депрессия, изменения в поведении), прогрессирующим мозжечковым синдромом, миоклонусом, другими неврологическими симптомами. В отличие от спорадической формы, новый вариант типичен медленным развитием вирусной инфекции; прионная болезнь затрагивает более молодые возрастные группы. Передача происходит алиментарным путем (при потреблении мяса пораженных животных). Инкубационный период – более 10 лет. В мире на сегодняшний день погибло около 200 человек.
- Детский вариант болезни Крейцфельдта-Якоба – болезнь Альпера. Имеет те же проявления, что и заболевание у взрослых; в дополнение сопровождается стеатозом печени.
Болезнь Герстманна-Штрюсслера-Шейнкера (Gerstmann-Sträussler-Scheinker disease)
Болезнь Герстманна-Штрюсслера-Шейнкера – это очень редкое аутосомно-доминантное наследственное заболевание, начинающееся в 3-4-ой декаде жизни. Проявляется медленным замедлением работы нейронов прионами, прогрессирующей дисфункцией мозжечка и спинного мозга. Признаки:
Свойства молекул
Трехмерные структуры С-концевых участков белков PrP
C
(слева) и PrP
Sc
. Фиолетовым цветом окрашены альфа-спирали, зелёным — бета тяжи.
N-концевой участок белка релаксирован и не поддаётся рентгеноструктурному анализу.
https://www.youtube.com/watch?v=i1pt_
Прионовые белки млекопитающих не сходны с прионовыми белками дрожжей по аминокислотной последовательности. Несмотря на это, основные структурные особенности (формирование амилоидных волокон и высокая специфичность, препятствующая передаче прионов от одного вида организмов к другому) у них общие. Вместе с тем, прион, отвечающий за коровье бешенство, обладает способностью передаваться от вида к виду.
Правый рисунок — модель двух конформаций приона; слева известная, нормальная, конформация структуры терминального участка C-terminal PrPC. (для отображения/загрузки см. RCSB Protein Databank).
Заболевания животных
Риск заболеваний животных для людей очень спекулятивен (лучше всего проблематику характеризует связь BSE и нового варианта расстройства Крейцфельдта-Якобса). У животных расстройства проявляются агрессивностью, нарушением двигательных способностей:
- cпонгиоформная энцефалопатия крупного рогатого скота (Bovine spongiform encephalopathy – BSE);
- скрепи;
- хроническая болезнь истощения (Chronic wasting disease – CWD);
- войлочная спонгиоформная энцефалопатия;
- трансмиссивная энцефалопатия.
Исследования прионов дрожжей и др. микромицетов
https://www.youtube.com/watch?v=ytcopyrighten-GB
Прион-подобные белки, поведение которых подобно поведению PrP найдены в природных популяциях микромицетов и дрожжей. Исследования прионов дрожжей подтвердили гипотезу о том, что превращение белков в прионное состояние зависит только от белков. Было показано, что прионы, экстрагированные из клеток, могут служить «семенами» образования прионов в пробирке.
Одним из наиболее хорошо изученных белков, склонных к образованию прионов у дрожжей — фактор терминации трансляции (eRF3), который образует так назваемые PSI клетки. Такие клетки имеют изменёное физиологическое состояние и изменённый уровень выражения некоторых генов, что позволило выдвинуть гипотезу о том, что у дрожжей образование прионов может играть адаптативную роль[1] .
Диагностика
Диагностика прионных болезней основана на обнаружении когнитивных расстройств, их прогрессирующем развитии, и уже на промежуточной стадии развития – не специфических нарушений ЭЭГ, постепенно переходящих на специфические паттерны (burst suppression pattern) с миоклониями в клинической и ЭМГ картине, эпилептическими приступами.
В спинномозговой жидкости присутствует только белок 14-3-3.
При спорадической форме болезни Крейцфельдта-Якобса МРТ на взвешенном изображении Т2 демонстрирует гиперинтенсивность в базальном и хвостовом ядре. При новом варианте заболевания она присутствует в пульвинарном таламусе, имеет форму клюшки.
Тестирование ДНК поможет выявить генетические и семейные формы расстройств, что означает выявление мутации E200K.
Подтверждением клинико-диагностического заключения являются иммуногистохимическое и нейрогистологическое исследование ткани головного мозга.
- типичная нейроцитарная вакуолизация;
- спонгиоматозная дегенерация нейропилы;
- потеря нервных клеток;
- астроглиоз;
- наличия скрепи-ассоциированных фибрилл в пораженном мозге.
Этиология
Человек может заразиться прионами, содержащимися в пище, так как они не разрушаются ферментами пищеварительного тракта. Беспрепятственно проникая через стенку тонкого кишечника, они в конечном итоге попадают в центральную нервную систему. Так переносится новый вариант болезни Крейтцфельдта-Якоба (nvCJD), которой люди заражаются после употребления в пищу говядины, содержащей нервную ткань из голов скота, больных бычьей губчатой энцефалопатией (BSE, коровье бешенство).
Прионы могут проникать в тело и парентеральным путем. Были описаны случаи заражения при внутримышечном введении препаратов, изготовленных из человеческих гипофизов (главным образом гормоны роста для лечения карликовости), а также заражение мозга инструментами при нейрохирургических операциях, поскольку прионы устойчивы к применяемым в настоящее время термическим и химическим методам стерилизации. Эта форма болезни Крейтцфельдта-Якоба обозначается как ятрогенная (1CJD).
При определённых, неизвестных условиях, в организме человека может произойти спонтанная трансформация прионового протеина в прион. Так возникает так называемая спорадическая болезнь Крейтцфельдта-Якоба (sCJD), впервые описанная в 1920 г. независимо друг от друга Гансом Герхардом Крейтцфельдтом и Альфонсом Марией Якобом.
Предполагается, что спонтанное возникновение этой болезни связано с фактом, что в норме в человеческом теле постоянно возникает небольшое количество прионов, которые эффективно ликвидируются клеточным Аппаратом Гольджи. Нарушение этой способности «самоочищения» клеток может привести к повышению уровня прионов выше допустимой границы нормы и к их дальнейшему неконтролируемому распространению. Причиной возникновения спорадической болезни Крейтцфельдта-Якоба согласно этой теории является нарушение функции Аппарата Гольджи в клетках.
Особую группу прионовых заболеваний представляют собой наследственные (врожденные) болезни, вызванные мутацией гена прионового протеина, который делает возникший прионовый протеин более подверженным спонтанному изменению пространственной конфигурации и превращения их в прионы. К этой группе наследственных заболеваний относится и наследственная форма болезни Крейтцфельдта-Якоба (fCJD), которая наблюдается в ряде стран мира.
При прионовой патологии наивысшая концентрация прионов обнаружена в нервной ткани заражённых людей. Значительное количество прионов встречается в лимфатической ткани. Наличие прионов в биологических жидкостях, включая слюну, пока не было однозначно подтверждено. Если представление о постоянном возникновении небольшого количества прионов верно, то можно предположить, что новые, более чувствительные методы диагностики откроют это количество прионов, разбросанное по различным тканям. В данном случае, однако, речь пойдёт о «физиологическом» уровне прионов, которые не представляют собой никакой угрозы для человека.
Как замедлить течение прионной болезни
До недавнего времени было сложно замедлить прионную болезнь, не говоря о ее излечении.
Современная нейрохирургия может облегчить дегенеративные осложнения путем пересадки нейронов при прионом заболевании.
Революционная попытка остановить патогенные протеины была впервые предпринята командой врачей из Университета Питтсбурга. Команда д-ра Кондзиолки в рамках исследования пересадила 12 пациентам нейроны in vitro (2-6 млн. LBS-нейронов). Через 12 месяцев после операции ни у одного из реципиентов не было зарегистрировано нежелательных последствий трансплантации, у всех новые клетки были приняты. У 6 пациентов произошло значительное улучшение состояния.
Несколько лет журнал «Nature» сообщил о разработке антител против прионов в Англии командой ученых во главе с Симоной Хоук (Imperial College). В тестах на зараженных мышах, грызуны, получившие антитела, были вылечены уже через месяц после заражения.
Врачи намерены использовать эту возможность очистить нейроны от патогенных протеинов для лечения нового варианта болезни Крейтцфельда-Якобса у людей. Дальнейшие исследования должны помочь улучшить лекарства, которые смогут очистить мозг и его микроглии от прионов.
Недавно М. Пфайфер и несколько других немецких ученых обнаружили и подтвердили способ, позволяющий очистить мозг от прионов. Они доказали, что РНК-интерференция может успешно лечить заболевания путем удаления из РНК-клетки части, ответственной за выработку патогенных белков. Это предотвратило их мутацию в опасные протеины.
Подобный метод – воздействие искусственно синтезируемых праймеров (олигонуклеотидов) против прионов. Как и РНК-интерференция, конструирование праймеров находится на стадии исследования.
Теоретически перспективным в лечении болезней может быть пептид PrP13, аналогичный структурному продукту, встречающемуся при этих заболеваниях. Вещество нацелено на ген PRNP, что должно повлиять на превращение PrPC в PrPres. Последняя названная патогенная молекула изменяется в структуру, подобную PrPC. Вещество используется под аббревиатурой PrP13, а его исследования на животных смогли продлить выживаемость на 50-300%.
Что же такое прионы и каков их механизм действия на организм (современные представления)?
На самом деле в организме человека и многих других живых существ есть белки PrPC. По-русски – нормальная форма прионных белков (открыты были после исследований Сиггурдсона, поэтому такая странность в название). Известна его длина, последовательность аминокислот, вторичная структура. Важно знать, что конечная структура состоит из трёх α-спиралей и двухцепочечного антипараллельного β-листа.
Обладают интересным свойством, а именно осаждаются высокоскоростным центрифугированием, что является стандартным тестом на наличие прионов. Есть данные, что PrP играет важную роль в прикреплении клеток, передаче внутриклеточных сигналов, а потому может быть вовлечён в коммуникацию клеток мозга. Тем не менее, функции PrP исследованы недостаточно.
(a) норма (b) патология
Эксперименты на мышах, лишенных этих белков, показывают, что отсутствие PrP приводит к демиелинизации нервов. Возможно, прионые белки в норме поддерживают долговременную память.
Но это в норме.
Иногда случаются «проблемы» и появляются белки, названные — PrPSc — инфекционные прионы. Отличаются они тем, что в них вместо α-спиралей преобладают β-слои.
Приводит это к тому, что меняется взаимодействие других белков с новым белком.Полбеды, если бы образовывался всего один белок на организм. Беда в том, что, однажды образовавшись, белок (!) сам начинает менять структуру других.Рассмотрим основные механизмы размножения PrPSc
Считается, что прионное заболевание может быть приобретено 3 путями: в случае прямого заражения, наследственно или спорадически (спонтанно) или их комбинациями.
Спорадическая (то есть спонтанная) прионная болезнь возникает в популяции у случайной особи. Таков, например, классический вариант болезни Крейтцфельдта — Якоба. Существуют две основные гипотезы относительно спонтанного появления прионных болезней. Согласно первой из них спонтанное изменение происходит в самом доселе нормальном белке в мозге, то есть имеет место посттрансляционная модификация.
Альтернативная гипотеза гласит, что одна или несколько клеток организма в какой-то момент претерпевают соматическую мутацию (то есть, не передающуюся наследственно) и начинают производить дефектный белок PrPSc. Как бы то ни было, конкретный механизм спонтанного возникновения прионных болезней неизвестен.
Вторая – заражение. По данным современных исследований, основной путь приобретения прионных заболеваний — поедание заражённой пищи. Считается, что прионы могут оставаться в окружающей среде в останках мёртвых животных, а также присутствуют в моче, слюне и других жидкостях и тканях тела (кровь, ликвор).
Из-за этого заражение прионами может произойти и в ходе пользования нестерильными хирургическими инструментами. Это усложняет стерилизацию хирургических инструментов или устройств на скотобойне. Прионы в большинстве своём устойчивы к протеазам, высокой температуре, радиации и хранению в формалине, хотя эти меры и снижают их способность к заражению.
Эффективная дезинфекция против прионов должна включать гидролиз или повреждение/разрушение их третичной структуры. Это можно достичь обработкой хлорной известью, гидроксидом натрия и сильнокислыми моющими веществами. Пребывание в течение 18 минут при температуре 134 °C в герметичном паровом автоклаве не может деактивировать прионы.
В качестве основного современного метода для деактивации и денатурации прионов в настоящее время изучается озоновая стерилизация. Ренатурация полностью денатурированного приона до инфективного состояния зафиксирована не была, однако для частично денатурированных прионов в некоторых искусственных условиях это возможно.
Еще стоит помнить, что эти белки могут долго сохраняться в почве за счёт связывания с глиной и другими почвенными минералами. Не впадайте в паранойю, но теоретически они могут быть повсюду.
https://www.youtube.com/watch?v=userprightcom
В 2011 году было сообщено об открытии прионов, передающихся по воздуху в частицах аэрозоля (то есть воздушно-капельным путём). Также в 2011 году было опубликовано предварительное доказательство того, что прионы могут передаваться с получаемым из мочи человеческим менопаузальным гонадотропином, применяемым для лечения бесплодия.
Теоретически с помощью всего одного больного животного с прионной болезнью, можно уничтожать целые нации и страны, просто добавляя его костную муку в кормовые добавки и продавая их в нужное государство.Сходная ситуация произошла в конце 80-х годов в Британии (эпидемия коровьего бешенства). Тогда, скорее всего по незнанию (а не по злому умыслу) произошел вышеуказанный процесс, унесший жизни около 200 человек (на 2009 год) и 179 тыс. голов крупного рогатого скота.
Размножение прионов
Третий механизм – генетический. Был открыт недавно и совершенно не вписывается в общую картину. Был идентифицирован ген, кодирующий нормальный белок PrP — PRNP, локализованный на 20-й хромосоме. При всех наследственных прионных заболеваниях имеет место мутация этого гена.
Попавший тем или любым образом «искаженный» прионный белок начинает изменять структуры близким к нему по структуре белков, превращая их в такие же патогенные агенты.
Основная гипотеза, наиболее близко отображающая этот процесс очень проста. Одна молекула PrPSc присоединяется к одной молекуле PrPC и катализирует её переход в прионную форму. Две молекулы PrPSc после этого расходятся и продолжают превращать другие PrPC в PrPSc.
Но схема дает больше вопросов, чем ответов.