Neuromuscular diseases
This is a group of hereditary diseases that are transmitted at the genetic level (X-linked, autosomal dominant, autosomal recessive forms, maternally inherited mitochondrial forms). About a third of disease cases are the result of new mutations. Taking into account the structural organization, the following levels of damage to the neuromuscular system are distinguished:
- Primary muscular lesions (muscular dystrophy of Duchenne, Becker, myopathy, myotonia).
- Synaptic lesions (neuromuscular transmission disorders - myasthenia gravis).
- Neuronal lesions (spinal muscular atrophy).
What is important to know for congenital muscular dystrophies/myopathies?
- Family Guide to Nursing Care for Congenital Myopathy (if the guide is not shown in the box below, you can view it at https://goo.gl/woCsw6):
- Family Guide to Nursing Care for Congenital Muscular Dystrophy (If the guide is not shown in the box below, you can view it at https://goo.gl/YKGySX
- For physicians, Consensus Statement on Standard Treatment for Congenital Myopathies (if the guideline is not shown in the box below, you can view it at https://goo.gl/sV4B1B):
SYMPTOMS OF NEUROMUSCULAR DISEASES
The main symptom of neuromuscular diseases is muscle weakness. The clinical picture depends on the affected area (shoulder girdle, hips, pelvis, lower extremities, facial muscles, respiratory muscles). Depending on the form of the disease, muscle weakness and increased fatigue during physical activity develop, muscle atrophy / pseudohypertrophy, fasciculations appear, tendon reflexes are inhibited , gait is disturbed, special motor techniques appear when getting up from a lying or sitting position. Neuromuscular diseases occur in stages. A neurologist will help determine the stage of development of pathological processes using medical diagnostics.
Hereditary paroxysmal myoplegia
Paroxysmal myoplegia is a rare hereditary disease in children. Characterized by periodic attacks of paralysis of the skeletal muscles. It is inherited according to the principle in which one mutant allele localized in the autosome is sufficient for the manifestation of the disease.
There are three forms:
- hyperkalemic;
- hypokalemic;
- normokalemic.
The hypokalemic form of paroxysmal myoplegia most often affects men.
The first attacks of the disease appear at the age of 10.
The attack occurs in the morning or at night. In this case, weakness is felt in the limbs and neck, often reaching paralysis. In some cases, paralysis spreads to the facial muscles and respiratory tract.
The attack is accompanied by thirst, hyperemia, and sweating. The duration can be from an hour to seven days. Women usually experience an attack on the first day of their period.
The hyperkalemic form can be found less frequently; its attacks begin at a young age. The attack is provoked by cold and prolonged muscle inactivity. It begins with a sensitivity disorder in the facial muscles, arms and legs.
The third form of the disease manifests itself before the age of 10. The attacks go away within a few days or weeks. They can be triggered by low ambient temperature, alcohol consumption, and heavy physical activity.
DIAGNOSTICS
The diagnostic task is to establish the level of damage to the neuromuscular system, establish the cause and determine whether there is a specific treatment.
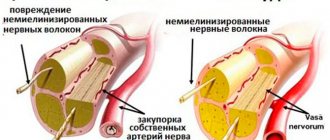
- Electroneuromyography and electromyography. Diagnostic methods help determine primary and secondary myopathy. Also assess the degree of damage to the spinal cord and peripheral nerves.
- Laboratory tests. A clinical and biochemical blood test is required to determine the level of electrolytes, protein, lactate, CPK, ALT, AST, myoglobin and thyroid hormones.
- Muscle biopsy with pathomorphological examination of muscle tissue. The diagnostic results help to establish an accurate diagnosis (Duchenne, Becker muscular dystrophy), which is important for selecting the correct therapy.
- Molecular genetic diagnostics is the most informative diagnostic method, which is prescribed for neuromuscular diseases.
- Magnetic resonance imaging of the brain and cervical spine.
- An electrocardiogram (ECG) evaluates heart rhythm.
- ECHO CG reveals hypertrophic, dilated cardiomyopathy.
Myotonia
Myotonia is a delayed relaxation of a muscle after tension.
The disease is divided into three types:
- action myotonia;
- percussion myotonia;
- electromyographic myotonia.
If a patient with action myotonia clenches his fingers into a fist and then tries to quickly straighten them, he will need time to fully straighten his palm.
The second type is characterized by muscle contraction during intense tapping with a hammer.
To detect electromyographic myotonia, a needle shock will be required. At the same time, the device shows high-frequency discharges, which are at first more frequent and then decrease in frequency.
TREATMENT OF NEUROMUSCULAR DISEASES
As for treatment, there is no specific therapy. The therapeutic approach is based on creating the prerequisites for successful rehabilitation and providing high quality general and specialized care.
- Drug therapy. The goal is to compensate for the energy deficiency of muscle tissue, improve tissue metabolism and peripheral circulation. Myotropic drugs and anabolic steroids are used. In the case of Duchenne muscular dystrophy, glucocorticosteroids are used. In the case of myasthenia gravis, anticholinesterase drugs are used. For secondary damage and/or dysfunction of the central nervous system, courses of neuroprotective and neurotrophic therapy are carried out. Drug therapy for bulbar and pseudobulbar disorders.
- Physiotherapy. The goal is to correct metabolic disorders, improve neuromuscular conduction, normalize muscle tone, and prevent the development of contractures.
- Physiotherapy. The goal is to prevent the development and correction of contractures of the limbs, maintain motor activity, improve muscle tone, delay the development of atrophies, and prevent complications caused by inactivity. Teaching the patient how to use assisted sitting and standing devices.
- Orthopedic correction.
- Early detection of swallowing disorders (dysphagia) and the possibility of drug correction (together with ENT doctors, based on the results of nasopharyngoscopy - the functional state of the epiglottis, the presence of regurgitation, and in older children + analysis of the results of an X-ray contrast study of the act of swallowing).
- Neurological assessment of respiratory function (ENMG analysis of the phrenic nerves + ultrasound of the diaphragm, analysis of respiratory function, pulse oximetry and capnometry, mechanics and type of breathing, ECHO data, ECG with a cardiologist’s conclusion).
Neurological examination of patients for respiratory failure makes it possible to clarify its causes, reversibility of disorders and the degree of their compensation. In the case of an established dependence on mechanical ventilation, work in a team of doctors to create a multifunctional therapeutic program that is aimed at improving physical condition and restoring normal life functions. choice of treatment methods and their order (medication and non-medication, including management of respiratory support parameters, breathing exercises, physical therapy, physiotherapeutic techniques, psychological assistance, etc.).
Myasthenia gravis
Myasthenia gravis is a disease that causes disruption of neuromuscular transmission and is manifested by weakness and pathological fatigue of skeletal muscles.
Etiology and pathogenesis.
Acquired myasthenia gravis is associated with the formation of antibodies against acetylcholine receptors on the postsynaptic membrane of the neuromuscular junction, which block the transmission of excitation from nerves to muscles. The thymus gland (thymus) appears to play an active role in the pathogenesis of the autoimmune reaction, but the reasons for its development remain unclear. The much rarer congenital myasthenia gravis is caused by a genetically determined defect of neuromuscular synapses. Neonatal myasthenia gravis is a transient condition observed in infants born to mothers with myasthenia gravis and is caused by the passage of maternal antibodies to acetylcholine receptors across the placenta.
Clinical picture.
Myasthenia gravis can occur at any age, but the highest incidence is observed in two age categories: from 20 to 40 and from 65 to 75 years. In the vast majority of cases, the disease primarily involves the eye muscles, so initially patients complain of occasional drooping of the eyelid and double vision. In the next 1-2 years, in most patients, the process involves the facial and bulbar muscles, muscles of the neck, limbs and trunk with the development of a generalized form of the disease. But in some patients the disease does not spread beyond the external muscles of the eye (ocular form). Characterized by pronounced fluctuations in symptoms during the day, in connection with this the disease is often mistaken for hysteria. The phenomenon of pathological muscle fatigue is manifested by an increase in symptoms during physical activity (for example, increased weakness of the masticatory muscles during eating, weakening of the voice during a conversation). After rest, symptoms decrease. Characterized by the absence of autonomic disorders (impaired innervation of the pupil or pelvic disorders), muscle atrophy, decreased tendon reflexes, and sensitivity disorders. A neurological examination reveals a decrease in strength that increases with repetition of movements. To identify pathological fatigue of the muscle that lifts the upper eyelid, the patient is asked to fix his gaze, looking up, to identify weakness of the muscles of the shoulder girdle - raise his arms up for 30-60 s, to detect fatigue of the muscles of the larynx - count out loud to 100. Selective involvement of muscles is characteristic. (for example, the neck flexors are weaker than the extensors), which makes it possible to distinguish myasthenia gravis from asthenia or hysteria. Patients with generalized myasthenia sometimes experience a rapid deterioration of their condition with the development of respiratory failure associated with weakness of the respiratory muscles or bulbar muscles (myasthenic crisis). A crisis can occur due to an unfavorable course of the disease (sometimes it is the first manifestation of myasthenia gravis), against the background of infection, electrolyte disturbances (hypokalemia, hypermagnesemia) or taking drugs that disrupt neuromuscular transmission. Severe respiratory failure during a crisis can develop very quickly, within a few minutes. Its approach is indicated by shortness of breath, inability to swallow saliva and keep the head straight, and weakening of the voice. Less commonly, an increase in muscle weakness and respiratory failure is caused by an overdose of anticholinergic drugs (cholinergic crisis). This version of the crisis is supported mainly by autonomic disorders associated with the activation of acetylcholine receptors: narrow pupils and accommodation paresis, hypersecretion of saliva and bronchial mucus, intestinal colic, diarrhea, vomiting, bradycardia, as well as generalized muscle twitching. But in some patients it is almost impossible to clinically differentiate a myasthenic crisis from a cholinergic one.
Diagnostics.
To confirm the diagnosis of myasthenia gravis, a proserine test is performed with 2 ml of a 0.5% solution of proserine, which is administered subcutaneously, and the effect is observed for 40 minutes. In patients with myasthenia gravis, there is a significant decrease and sometimes complete disappearance of the symptoms of the disease. To correct the possible side effects of proserin: bradycardia, bronchospasm, arterial hypotension, you should also have a syringe with 0.5-1 ml of 0.1% atropine solution and an Ambu bag ready. When administering prozerin, other undesirable effects are possible - hypersalivation, lacrimation, muscle twitching, diarrhea, intestinal colic, nausea, urinary and fecal incontinence. The diagnosis is also confirmed using electromyography, determining the content of antibodies to acetylcholine receptors. In adults with a confirmed diagnosis of myasthenia gravis, computed tomography of the chest is indicated to exclude tumors or thymic hyperplasia, which are detected in a significant proportion of patients.
Treatment.
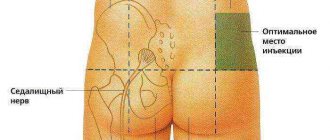
To reduce weakness and pathological fatigue of muscles, anticholine esterase agents are primarily used that inhibit the breakdown of acetylcholine in the synapse - pyridostigmine (kalimin) and neostigmine (prozerin). The effect of kalimine begins 30-60 minutes after taking the drug and lasts 3-6 hours. Treatment begins with 30 mg 3 times a day, then the dose is increased to 60-120 mg 4 times a day. Although pyridostigmine is effective in most patients, only a small proportion of them experience complete remission of symptoms. The effect of the drug on different muscles is different: for some, its dose may be insufficient, for others it may be excessive. Further increase in dose may increase weakness in the latter case. To avoid overdose, the next dose should be taken no earlier than signs of the end of the previous dose appear. Frequent side effects are abdominal pain, nausea, diarrhea, and hypersalivation. Sometimes atropine (0.5 mg orally) is prescribed to reduce them, but it is not possible to take it regularly due to its toxic effect (nevertheless, it is useful for patients to have atropine with them). Adverse reactions can be reduced by reducing the single dose of anticholinesterase drugs, increasing the frequency of administration, or taking the drug with food. Prozerin has a shorter duration of action. It is often prescribed orally (15-30 mg) or parenterally (0.5-1.5 mg) to obtain short-term additional effect, for example, before lunch. At the same time, patients are often prescribed potassium supplements. If anticholinesterase drugs are insufficiently effective, corticosteroids are prescribed. They cause improvement in 70% of patients, but in the first 3 weeks, especially if treatment is started with a high dose, muscle weakness (including bulbar and respiratory) may increase. When a stable effect is achieved, begin to slowly reduce the dose. In a significant proportion of cases, patients are forced to take a maintenance dose of the drug for many years. In severe cases, when corticosteroids are poorly tolerated, immunosuppressants (azathioprine, less commonly cyclosporine and cyclophosphamide) are prescribed. Removal of the thymus gland (thymectomy) is indicated for patients under 60 years of age with a generalized form of the disease, as well as in the presence of a tumor of the thymus gland (thymoma). When caring for a patient with myasthenia gravis, the nurse should assist them in maintaining a hygienic regime, nutrition (special care is necessary if swallowing is impaired), and monitor the state of motor and respiratory functions. Medicines should be dispensed strictly as prescribed by the doctor. A number of drugs can increase the symptoms of myasthenia gravis, including some antibacterial agents (streptomycin, gentamicin and other aminoglycosides, tetracycline, ampicillin, erythromycin, ciprofloxacin, clindamycin, sulfonamides), beta blockers, lidocaine, quinine, procainamide, calcium antagonists, antiepileptics drugs (difenin, carbamazepine, barbiturates), aminazine, amitriptyline, diazepam (Relanium) and other benzodiazepines, muscle relaxants, diuretics (with the exception of potassium-sparing), magnesium salts, etc. If signs of a myasthenic crisis appear, the patient should be urgently hospitalized in the intensive care unit . It is better to carry out transportation in a half-sitting position. During transportation, first of all, care should be taken to ensure airway patency and prevent aspiration; it is necessary to remove mucus from the throat and give oxygen (through a mask or nasal catheter). Sometimes intubation is indicated. In the absence of signs of an overdose of anticholinesterase drugs (!), 1 - 2 ml of a 0.05% solution of proserine can be administered subcutaneously. Intravenous administration of the drug gives a faster effect, but is fraught with the risk of cardiac arrest, so it is used only in the most severe cases. Preliminarily, 0.5 ml of a 0.1% atropine solution is administered intravenously or subcutaneously. Further administration of proserin is possible only if a positive result is obtained from the first administration. In the intensive care unit, regular monitoring of the state of respiratory function and airway patency is established. If respiratory failure develops, intubation is performed and artificial ventilation is started. Electrolyte disturbances are corrected. If there are signs of infection, antibiotics are prescribed (preferably cyclosporine). Patients are often agitated, but sedatives generally should not be administered, as many of them worsen muscle weakness. Encouraging words and efficiency of the staff often sufficiently calm the patient; in severe cases, haloperidol is administered (1 ml of 0.5% solution intravenously or intramuscularly). Plasmapheresis gives the best results during a crisis. Sometimes during a crisis, corticosteroids are also used (for example, prednisolone, up to 100 mg/day orally), but an initial increase in weakness and respiratory failure is possible. If the cholinergic component of the crisis is reliably excluded, then in the absence of the need for artificial ventilation, the administration of proserin is continued. With the start of artificial pulmonary ventilation, which is usually carried out for 3-6 days, prozerin is canceled or its dose is reduced by half. In case of a cholinergic crisis, anticholinesterase drugs are temporarily discontinued, airway patency is restored, atropine is administered subcutaneously (0.5-1 ml of 0.1% solution every 2 hours) until dry mouth appears, cholinesterase reactivators are prescribed, if necessary, intubation and artificial ventilation of the lungs.
AP propagates through activation of Na + channels to nerve endings, where it depolarizes the cell membrane, which leads to the opening of voltage-gated Ca 2+ channels. Ca 2+ ions entering the nerve endings trigger the release of vesicles containing ACh from the presynaptic membrane, as a result of which the latter is released into the synaptic cleft. ACh then binds to receptors on the subsynaptic membrane and opens nonspecific cation channels. Depolarization of the subsynaptic membrane spreads to the postsynaptic membrane, where, after the opening of voltage-gated Na + channels, an AP occurs, which quickly spreads throughout the muscle membrane. ACh is destroyed by acetylcholinesterase, the resulting choline is recaptured by the nerve ending and reused for the synthesis of ACh.
Pathological changes can affect any element of this process. Local anesthetics, for example, inhibit voltage-gated Na + channels of neurons, thereby disrupting nerve transmission to the end plate of the neuromuscular junction. Ca 2+ channels can be blocked by antibodies. Botulinum toxin inactivates the synaptobrevin protein, which is responsible for the binding of vesicles containing ACh to the plasma membrane, i.e., for the release of ACh. Acetylcholine receptors, as well as Ca 2+ channels, can be blocked by antibodies, which, in addition, accelerate internalization and destruction of these receptors. Receptors can also be blocked by curare, which, without having its own effect, competitively inhibits the binding of ACh to receptors.
Succinylcholine (suxamethonium chloride) leads to prolonged stimulation of receptors, prolonged depolarization of the postsynaptic membrane, thereby causing inactivation of postsynaptic Na + channels. Thanks to this action, it is capable, like curare, of blocking neuromuscular transmission of impulses. At low concentrations, acetylcholinesterase inhibitors (eg, physostigmine) facilitate neuromuscular transmission by increasing the availability of ACh at the synaptic cleft. However, in high doses they slow down neuromuscular transmission, because high concentrations of ACh and succinylcholine cause prolonged depolarization of the subsynaptic membrane, thereby inactivating postsynaptic Na + channels. Reuptake of choline by nerve endings may inhibit Mg 2+ ions and hemicholine.
The most important disease that affects the end plates of neuromuscular junctions is myasthenia gravis, characterized by muscle paralysis due to blockade of neuromuscular impulse transmission. This disease is caused by the formation of antibodies to ACh receptors on the subsynaptic membrane, accelerating the destruction of these receptors. This autoimmune disease can be triggered by viral infections, which stimulate the expression of MHC molecules, which facilitates the recognition of the antigen by the immune system. Myasthenia gravis can also occur in patients with benign thymic tumors. The formation of such autoantibodies more often occurs in individuals who are carriers of specific subtypes (DR3 and DQw 2) of MHC class II or HLA. In rare cases, myasthenia gravis is caused by genetic defects in the ACh receptor or acetylcholinesterase channels. In patients suffering from myasthenia gravis, repeated stimulation of the motor nerves will initially cause the formation of normal summed action forces in the muscles, the amplitude of which, however, will decrease due to the progressive increase in “fatigue” of neuromuscular transmission.
Another immune autoaggressive disease in which neuromuscular transmission is disrupted is Lambert-Eaton pseudomyasthenia syndrome. This condition often develops in patients with small cell lung cancer. Ca 2+ channels in the plasma membrane of tumor cells sensitize the immune system and stimulate the formation of antibodies, which also interact with the Ca 2+ channels of the end plates of neuromuscular synapses. Due to the inhibition of Ca 2+ channels, the summed muscle action potential is initially small, but then it gradually normalizes, since repeated stimulation increases the amount of Ca 2+ accumulating in the nerve endings.
Acetylcholine is secreted by motor nerve endings not only during excitement, but also at rest. The only difference is that at rest small portions - “quanta” - of acetylcholine are released, and under the influence of a nerve impulse a significant number of such “quanta” are simultaneously released into the synaptic cleft. “Quantum” is a “package” of transmitter molecules in a single vesicle of a nerve ending, pouring its contents into the synaptic cleft. In the end plate of various animals, each “quanta” contains up to 2000 molecules of acetylcholine. The release of individual quanta into the synaptic cleft at rest causes a short-term weak depolarization of the postsynaptic membrane of the muscle fiber. This depolarization is called a miniature potential, since its amplitude (0.5 mV) is 50-80 times less than the EPP caused by a single nerve impulse. Miniature potentials usually occur with a frequency of approximately one per second; they are recorded not only at neuromuscular junctions, but also at the synapses of nerve cells in the central nervous system.
Congenital non-progressive myopathies
In most cases, the area of the lower extremities is affected, less often - the upper extremities; in exceptional cases, damage to the cranial muscles occurs - impaired facial expressions and eye movements.
During the development and growth of a child, problems with motor skills are noted; children often fall, begin to sit and walk late, and cannot run or jump. There are no intellectual impairments. Unfortunately, this type of myopathy is incurable.

Myopathy
The term myopathy (myodystrophy) unites a fairly large group of diseases that are united by a common feature: primary damage to muscle tissue. The development of myopathy can be triggered by various factors: heredity, viral infection, metabolic disorders and a number of others.
Inflammatory myopathies (myositis) include diseases caused by the inflammatory process. They develop as a result of autoimmune disorders and may be accompanied by other diseases of a similar nature. These are deramtomysitis, polymyositis, myositis with various inclusions.
Mitochondrial myopathies. The cause of the disease is structural or biochemical mitochondria. This type of disease includes:
In addition to these diseases, there are a number of rare types of myopathies that affect the central core, endocrine system, etc.
When active, myopathy can lead to disability and further immobilization of the patient.