| Наследственная оптическая нейропатия Лебера | |
| МКБ-10 | 47.247.2 |
| МКБ-10-КМ | H47.22 и H47.2 |
| МКБ-9 | 377.16377.16 |
| OMIM | 535000 |
| DiseasesDB | 7340 |
| MeSH | D029242 |
| Медиафайлы на Викискладе | |
Наследственная оптическая нейропатия LHON Лебера
, или
атрофия зрительного нерва Лебера
, является наследственной (передаётся от матери к потомству) митохондриальной дегенерацией ганглионарных клеток (РСК) сетчатки и их аксонов, что приводит к острой или почти острой потере центрального зрения; это влияет преимущественно на молодых мужчин. Тем не менее, LHON передается только по материнской линии, прежде всего, из-за мутаций (не ядерных) в митохондриальном геноме, и только яйцеклетка способствует митохондрии в зародыше. LHON, как правило, связана с одной из трех патогенных митохондриальных ДНК (мтДНК) точечных мутаций. Эти мутации действуют на нуклеотиды и репозиционируют 11778 в , 3460 в и 14484 в , соответственно, в ND4, ND1 и Nd6 субъединицах генов в комплексе I окислительного фосфорилирования цепочек в митохондриях. Мужчины не могут передать болезнь своему потомству.[1]
Причины возникновения заболевания, тип наследования
Недуг зачастую прогрессирует у пациентов в возрасте от 12 до 25 лет.
При этом весомую роль в возникновении заболевания играет
наследственный фактор
. По клиническим показателям болезнь схожа с двусторонним ретробульбарным невритом.
В течение двух дней
развивается внезапная потеря зрения на обоих глазах, иногда острота снижается сперва на одном, а потом и на другом органе.
В течение последующих двух недель
качество зрения продолжает падать, а затем останавливается на определённом уровне. Полная слепота появляется относительно редко.
Характерная особенность наследственной оптической нейропатии Лебера — неполная пенетрантность
(
до 40% у мужчин и 15% у женщин
) и высокая частота поражения среди мужчин (они заболевают
в 5 раз чаще
, чем женщины). Это, возможно, связано с воздействием Х-сцепленного модифицирующего гена, находящегося в районе
Xp21.
Наиболее частыми причинами развития недуга считаются:
- инфекционные воспаления ЦНС и зрительных нервов;
- врожденные и приобретенные гидроцефалические патологии;
- онкология черепной коробки;
- церебральный паралич;
- метаболические нарушения;
- интоксикации (свинцом, лекарственными препаратами, ртутью);
- врожденные и генетические патологии зрительного нерва.
Влияние на развитие заболевания оказывают следующие факторы:
- стресс;
- курение и употребление спиртных напитков;
- воздействие токсинов;
- некоторые лекарства и инфекции.
Эпидемиология
У населения Северной Европы примерно у одного из 9000 человек имеется один из трех основных видов мутаций LHON.[11][12]Распространённость заболевания в Европе составляет от 1: 30000 до 1: 50000.
Мутация LHON ND4 G11778A доминирует в качестве основной мутации в большинстве стран мира с 70 % случаев Северной Европы и 90 % случаев азиатских стран. Из-за эффекта основателя, мутация LHON T14484C ND6 происходит в 86 % случаев LHON в Квебеке,Канада .[13]
Более 50 процентов мужчин и более 85 процентов женщин с мутацией никогда не испытывают потерю зрения или связанных с ней медицинских проблем. Конкретный тип мутации может предсказать вероятность пенетрантности, тяжесть заболевания и вероятность восстановления зрения у пострадавших. Как правило, женщина, которая гомоплазмически вынашивает, основная LHON мутация имеет ~ 40 % риск появления больного сына и ~ 10 % риск появления больной дочери.
Дополнительные факторы могут определить, развиваются ли у человека признаки и симптомы этого расстройства. Экологические факторы, такие как курение и употребление алкоголя могут быть использованы, хотя исследования этих факторов, дали противоречивые результаты. Исследователи также изучают изменения дополнительных генов, в частности генов на Х-хромосоме,[14][15] их вклад в развитие признаков и симптомов. Степень гетероплазмии, процент митохондрий, которые имеют мутантные аллели, могут также играть определенную роль.[16] Модели митохондриальных аллелей называемых гаплогруппами также может влиять на экспрессию мутаций.[17]
Симптомы наследственной атрофии зрительного нерва Лебера
На начальных стадиях заболевания глазное дно остается без изменений, иногда отмечают лишь некоторую гиперемию сосочков зрительных нервов
и
размытие границ
. При диагностике полей зрения наблюдаются
центральные скотомы.
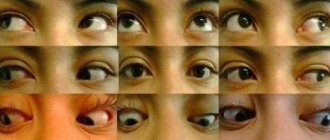
Фото 1. Так выглядит глазное дно при нормальном состоянии органа зрения (слева) и при атрофии зрительного нерва (справа).
Атрофию классифицируют на несколько типов:
- простой (первичный) и вторичный (поствоспалительный или послезастойный)
— первый характеризуется обычной потерей зрения, сужением бокового зрительного поля; - частичный и полный тип
— полная или частичная потеря зрения; - стационарный или прогрессирующий
— при первой разновидности процесс потери зрения на какой-то стадии останавливается, а при прогрессирующей форме наблюдается постепенный спад зрительной функции, который может привести к полной атрофии нерва, т. е. к слепоте; - односторонний и двусторонний тип
— поражение одного или двух глаз.
Справка.
У большинства пациентов обнаруживают прогрессивное ухудшение зрения в течение месяцев и даже лет, но примерно
у 20% больных
отмечают улучшение зрения. Известны случаи полного восстановления зрения.
Перечень симптомов атрофии зрительного нерва довольно обширен и зависит от вида патологии. Самые распространённые симптомы всех типов патологии:
- снижение остроты зрения;
- нарушение аккомодации;
- куриная слепота.
В запущенном состоянии
болезни к общим симптомам повреждения зрительных путей присоединяются признаки, свидетельствующие о поражении ЦНС.
Сюда относятся случаи:
- осложнённой деменции;
- депрессии;
- появления бульбарных симптомов;
- атаксии мозжечкового и спинального типа;
- спастической параплегии.
В таких ситуациях проводят дифференциальную диагностику,
чтобы исключить риск возникновения рассеянного склероза, опухоли зрительного нерва или хиазмальной области.
Внимание!
Заболевание развивается в молодом возрасте (чаще
от 12 до 25 лет
), поэтому любые признаки нарушения зрительной функции не должны оставаться без внимания.
Основные симптомы болезней глаз, требующие обращения к офтальмологу
- Постепенное или резкое ухудшение зрения при рассматривании предметов вдали, необходимость напряжения и прищуривания.
- Внезапно возникшая острая боль, которая слабеет при закрывании глаза – подозрение на поражение роговицы.
- Постоянное ощущение соринки в глазу.
- Покраснение глаз, приступообразные боли, боязнь света, белесоватые или гнойные выделения из глаз.
- Сильные головные боли, сопровождающиеся уменьшением поля зрения.
- Возникшая пелена перед глазами и значительная потеря зрения.
- Сухость глаз.
- Слезотечение.
- Появление «тумана» перед глазами.
- Нечеткое изображение предметов, которые ранее видели отчетливо и другие.
Методы диагностики
Симптомы атрофии зрительного нерва появляются не только на первых порах развития недуга, но и в результате получения серьезных травм участков головного мозга
, которые ответственны за зрительные функции.
При осмотре пациента врач изучает
наличие сопутствующих заболеваний, факты приема фармакологии и контакта с химическими препаратами, пагубные привычки, а также жалобы, которые свидетельствуют о возможных интракраниальных поражениях.
При физикальной форме диагностики
офтальмологи определяют присутствие либо отсутствие экзофтальма, исследуют подвижность глазного яблока, тестируют реакцию зрачка на свет, рефлекс роговицы. Обязательно проводят проверку остроты зрения, периметрию, изучение цветовосприятия.
Перечень диагностических мер:
- офтальмоскопия
— анализируется степень размытия границы нерва;
Фото 2. Процесс офтальмоскопии: специальный прибор направляет пучок света на глаз, что помогает увидеть глазное дно пациента.
- тест на остроту зрения
, определение границы зрительного поля; - ангиография
сосудов головного мозга, питающих нерв; - выявление повреждённых участков нерва при помощи компьютерной периметрии;
- томография;
- краниографическое исследование;
- ЗВП
, определяющее снижение лабильности и увеличение порога чувствительности зрительного нерва; - при глаукоме измеряют внутриглазное давление;
- обзорная рентгенография орбиты глаза
— изучение патологий глазницы; - флуоресцентная ангиография
— осмотр сосудистой сетки сетчатки; - рентгенография
черепа и турецкого седла; - магнитно-резонансная томография (МРТ)
— оценка волокон зрительного нерва; - анализ крови
, подтверждающий или опровергающий наличие воспалительного процесса; - ИФА и ПЦР-диагностика.
Лечите глазные болезни в ОН КЛИНИК!
В офтальмологии очень большое значение имеет правильная и своевременная диагностика. Наша клиника оснащена оборудованием от лучших мировых производителей, которое отличается высочайшей точностью результатов обследования и позволяет выявлять любые патологии на самых ранних стадиях, когда сам человек даже не подозревает о существовании проблем со зрением.
Огромный опыт и профессионализм наших офтальмологов, их ориентирование на передовые международные технологии, постоянное изучение новых тенденций в мире офтальмологии и успешное применение практических наработок зарубежных коллег позволяет возвращать нашим пациентам возможность хорошо видеть.
Лечение
Недуг опасен тем, что разрушенное нервное волокно не подлежит регенерации.
Эффект от терапии может быть обусловлен только восстановлением функционирования дееспособных на момент воздействия волокон.
Лечение подразумевает нормализацию кровообращения
и
стимулирование процессов жизнедеятельности в угнетенных нервных волокнах
. С этой целью применяются сосудорасширяющие лекарства, препараты, которые улучшают трофические свойства, а также стимулируют ЦНС.
Важно!
Лечение недуга более результативно при длительном нахождении лекарственного вещества в зоне повреждения. Для получения максимального эффекта
нужны многочисленные инъекции
, а это довольно болезненно.
Ирригационная терапия
позволит дробно вводить лекарственные средства. Медикаменты поступают через катетер, который устанавливается в ретробульбарное пространство через отверстие в коже в нижнем углу глазницы. Катетер закрывают стерильной пробкой и фиксируют пластырем к коже.
У маленьких детей процесс выполняют под ингаляционным масочным наркозом. У старших — под местным. Лекарственные препараты вводят 5—6 раз в день
при помощи прокалывания иглой шприца пробки катетера после предварительной обработки этиловым спиртом. Набор препаратов подбирает врач в зависимости от стадии недуга. Срок лечения
7—10 дней.
Описание:
Оптическая Лебера (атрофия зрительного нерва Лебера) — наследственное заболевание, характеризующееся быстро или постепенно развивающимися двусторонними нарушениями центрального зрения у соматически здоровых молодых людей.
История исследования проблемы. Заболевание впервые описано Theodor Leber в 1871 г., который сообщил о 15 пациентах из четырех семей. В последующем многие авторы опубликовали свои наблюдения за семьями из Европы, Азии, Америки, Африки и Австралии, у членов которых в нескольких поколениях выявляли оптическую нейропатию.
Причины наследственной оптической нейропатии Лебера:
Внимание большинства исследователей было сосредоточено на изучении механизмов наследования патологии. Хитросплетения проблемы исследования механизмов наследования оптической нейропатии Лебера заключаются в том, что заболевание передается исключительно по материнской линии и развивается преимущественно у лиц мужскою пола. В 1963 г. A. van Senus доказал, что оно никогда не передается мужчинами. В то же время при Х-сцепленных рецессивных заболеваниях ожидаемая частота поражения женщин не превышает 1 %, что значительно ниже уровня, наблюдаемого при оптической нейропатии Лебера. Основываясь на этих противоречивых фактах, D.C. Wallace (1970) сделал следующее заключение: «Поверхностное изучение пораженной семьи создает впечатление о сцепленном с полом наследовании. Исследователь, завершающий работу, может предполагать цитоплазматическое наследование. Более детальный анализ приводит к путанице». В своих последующих работах он подчеркнул важное значение внехромосомных материнских факторов. Некоторые авторы не исключали возможности переноса персистирующих вирусоподобных субстанций в материнскую овоплазму или трансплацентарного поражения эмбриона.
Одновременно изучалось влияние факторов внешней среды на течение заболевания. J. Wilson (1965) обнаружил корреляцию между тяжестью клинических проявлений оптической нейропатии Лебера и курением. На основании своих наблюдений он предположил, что в основе зрительных расстройств у пациентов с оптической нейропатией Лебера лежат нарушения метаболизма цианидов. Позднее ТА. Berninger и соавт. (1989) выявили повышение уровня цианидов в крови у пациентов с оптической нейропатией в острой фазе. К. Tsao и соавт. (1999) подтвердили корреляцию стажа и объема курения с пенетрантностью и экспрессивностью оптической нейропатии Лебера.
Генетические исследования. В настоящее время доказано, что развитие нейропатии Лебера обусловлено точковыми мутациями в митохондриальной ДНК, приводящими к замене одной аминокислоты другой. Предпола¬гаемое митохондриальное наследование оптической нейропатии Лебера было подтверждено в 1988 г. D.C Wallace и соавт., которые первыми идентифицировали точку мутации митоховдриальной ДНК в нуклеотвдной позиции 11778 у больных с оптической нейропатией Лебера из девяти семей. В результате этой мутации происходит замещение аргинина на гистидин в кодоне 340 гена субъединицы 4 НАДФ-дегидрогеназы. Мутация 11778 идентифицирована в 50-60 % всех обследованных генетиками семей, члены которых были больны нейропатией Лебера.
Другую патогенетически значимую (первичную) мутацию в положении 3460 митохондриальной ДНК у больных с нейропатией Лебера из трех семей, у членов которых не выявлена мутация 11778, установили К. Huoponen и соавт. (1991). Данная мутация в колоне 52 гена, кодирующего субъединицу 1 НАДФ-дегидрогеназы, приводит к замене аланина треонином. Позднее N. Howell и соавт. (1991), а также D.R. Johns (1992) идентифицировали мутацию в нуклеотидной позиции 3460 еще в 15 семьях больных с оптической нейропатией Лебера. Мутация в точке 3460 определена приблизительно у 8 % пациентов с нейропатией Лебера.
Вскоре бьыи обнаружены мутации в нуклеотидных позициях 15257 и 15182 в гене апоцитохрома b. При обследовании 120 семей, члены которых были больны оптической нейропатией Лебера, мутация 15257 выявлена у 8 % родословных.
D.Besch и соавт. (1999) установили у больного с оптической нейропатией Лебера, его матери и четырех непораженных членов этой семьи по материнской линии первичную точковую мутацию митохондриальной ДНК в нуклеотидной позиции 14568. Эта мутация обусловливала замену глицина на серии в гене ND6. У матери пробанда не обнаружено зрительных расстройств, но при выявлена перипапиллярная микроангиопатия.
Это далеко не весь перечень мутаций, установленных у больных с оптической нейропатией Лебера. В настоящее время известно около 20 точковых мутаций. Во всех случаях они затрагивают компоненты комплексов (и III дыхательной цепи окислительного фосфорилирования.
Комплекс 1 (NADH-дегидрогеназный комплекс) — самый крупный белковый компонент дыхательной цепи, содержащий более 22 полипептидных цепей, кодируемых как митохондриальным, так и ядерным геномами. Установлено, что мутация G3460A в гене ND1 в культивированных фибробластах приводит к снижению активности NADH-дегидрогеназы до 40 % по сравнению с нормой, при этом связанный с комплексом I синтез АТФ остается неизменным.
V.Carelli и соавт. (1999) исследовали влияние мутаций 14459 и 14484 в субьединице ND6 (Met 64 Val) на комплекс I. Мутация 14459 выявлена у больных с оптической нейропатией Лебера, сочетающейся с мышечной дистонией. Данная мутация индуцирует восстановление специфической активности NADH-дегидрогеназного комплекса и увеличивает его чувствительность к децилубихинолу — субстрату комплекса Ь-С| дыхательной цепи (комплекс II). Таким образом, комплекс I ингибируется продуктом катализируемой им реакции. Мутация 14484 не оказывает подобного влияния на специфическую активность комплекса I, однако обусловливает более высокую чувствительность этого ферментного комплекса по сравнению с немутированными формами к ингибиторам — аналогам убихинона.
Анализ 70 различных последовательностей ND6-субъединицы показал, что мутации 14484 подвержена наиболее консервативная область белковой молекулы, локально напоминающая участок цитохрома Ь, взаимодействующий с убихиноном или убихинолом в цитохромоксидазном комплексе (комплекс III).
Таким образом, все мутации воздействуют на систему окислительного фосфорилирования, нарушая тем самым энергетический обмен в клетке. Согласно гипотезе A. Sadun (1998), в результате уменьшения числа молекул АТФ ниже определенного порогового уровня блокируется антероградный аксональный транспорт митохондрий, что приводит к дефициту АТФ и в итоге к гибели нейрона.
Идентификация диагноза с помощью анализа митохондриальной ДНК показала, что оптическая нейропатия Лебера охватывает более широкий возрастной диапазон, а женщины болеют значительно чаще, чем предполагали раньше. Н. Thicme и соавт. (1999) сообщили о семье, у 9 членов которой была оптическая нейропатия Лебера, обусловленная мутацией 11778 субъединицы ND4, восемь из них — девочки в возрасте до 10 лет.
Пять мутаций (11778, 3460, 14484, 15257 и 14568) вместе составляют свыше 90 % точковых мутаций, выявленных во всех родословных с фенотипом оптической нейропатии Лебера. Предполагают, что каждая из них имеет первичное патогенетическое значение. Идентифицированы также несколько вторичных мутаций, которые, воздействуя сочетанию как синергисты, могут вызывать развитие оптической нейропатии Лебера.
R.I Oostra и соавт. (1994) исследовали распределение 7 мутаций митохондриальной ДНК и их связь с клиническими проявлениями оптической нейропатии Лебера у 334 пациентов из 29 семей. Мутации в нуклеотидных позициях 11778, 3460 и 14484, встречающиеся только при оптической нейропатии Лебера, были обнаружены в 15, 2 и 9 семьях соответственно. В 3 семьях не выявлена ни одна из этих мутаций. Мутации в нуклеотидных позициях 15257, 13708, 4917 и 4216, которые ранее были обнаружены как у пациентов с оптической нейропатией Лебера, так и у здоровых людей, идентифицированы в 1, 10, 3 и 12 семьях соответственно. Комбинации мутаций митохондриальной ДНК обнаружены у членов большинства обследованных семей. У пациентов из 11 семей, в которых была выявлена только одна мутация — 11778, заболевание манифестировало в среднем в возрасте 29,2 лет, а итоговая острота зрения составляла в среднем 0,113. Тяжесть фенотипических проявлений оптической нейропатии Лебера у больных с мутациями в других нуклеотидных позициях зависела от митохондриального генотипа.
В семьях больных с оптической нейропатией Лебера описано явление гетероплазмии: количество мутантной митоховдриальной ДНК варьирует у разных пациентов от 5 до 300 % от всей имеющейся митоховдриальной ДНК. Заболевание развивается в тех случаях, когда процентное содержание мутантной ДНК достигает порогового значения. Количество мутантной ДНК в клетках организма коррелирует с тяжестью клинических симптомов. В процессе эволюции гетероплазмия митохондриальной ДНК может эффективно поддерживаться на протяжении нескольких поколений. При этом у части лиц возможна фиксация одной из форм митохондриальной ДНК в результате случайной полной элиминации другой формы в ходе оогенеза. Так, в некоторых случаях благодаря гетероплазмии у матери мутантная ДНК не передается детям. М.Т. Lott и соавт. (1990) сообщили о родословных, в которых количество мутантной ДНК увеличивалось от поколения к поколению, коррелируя с тяжестью фенотипических проявлений. Авторы также обнаружили, что содержание мутантной ДНК в крови и волосах различно у одного и того же субъекта. Таким образом, выявление и количественная оценка гетероплазмии имеет чрезвычайно важное значение для диагностики, прогнозирования и определения фенотипической экспрессии.
Хотя атрофию зрительного нерва Лебера традиционно считают семейным наследственным заболеванием, в настоящее время достаточно часто клиницисты наблюдают больных, манифестацию заболевания у которых можно рассматривать как спорадическую. Семейные случаи оптической нейропатии Лебера составляют 43 % от общего числа пациентов для мутации 11778, 78 % — для мутации 3460, 65 % — для мутации 14484 и 57 % — для мутации 15257.
Примечания
- Bandelt H.J., Kong Q.P., Parson W., Salas A.
More evidence for non-maternal inheritance of mitochondrial DNA? (англ.) // Journal of Medical Genetics (англ.)русск. : journal. — 2005. — December (vol. 42, no. 12). — P. 957—960. — doi:10.1136/jmg.2005.033589. — PMID 15923271. - Leber T. Ueber hereditaere und congenital angelegte sehnervenleiden (1871) Graefes Arch Clin Exp Ophthalmol. 17:249-291
- Erickson R.P.
Leber’s optic atrophy, a possible example of maternal inheritance (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 1972. — Vol. 24, no. 3. — P. 348—349. - Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas LJ 2nd, Nikoskelainen E.K.
Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy (англ.) // Science : journal. — 1988. — Vol. 242, no. 4884. — P. 1427—1430. — doi:10.1126/science.3201231. — PMID 3201231. - Huoponen K., Vilkki J., Aula P., Nikoskelainen E.K., Savontaus M.L.
A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 1991. — Vol. 48, no. 6. — P. 1147—1153. - Johns D.R., Neufeld M.J., Park R.D.
An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy (англ.) // Biochemical and Biophysical Research Communications (англ.)русск. : journal. — 1992. — Vol. 187, no. 3. — P. 1551—1557. — doi:10.1016/0006-291x(92)90479-5. - Nikoskelainen E.K., Marttila R.J., Huoponen K., et al.
Leber’s «plus»: neurological abnormalities in patients with Leber’s hereditary optic neuropathy (англ.) // Journal of Neurology, Neurosurgery and Psychiatry (англ.)русск. : journal. — 1995. — August (vol. 59, no. 2). — P. 160—164. — doi:10.1136/jnnp.59.2.160. — PMID 7629530. - cardiac arrythmia
- Mayo Clinic: Multiple Sclerosis
- OMIM 535000
- Man P.Y., Griffiths P.G., Brown D.T., Howell N., Turnbull D.M., Chinnery P.F.
The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2003. — February (vol. 72, no. 2). — P. 333—339. — doi:10.1086/346066. — PMID 12518276. - Puomila A., Hamalainen P., Kivioja S., et al.
Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland (англ.) // European Journal of Human Genetics (англ.)русск. : journal. — 2007. — October (vol. 15, no. 10). — P. 1079—1089. — doi:10.1038/sj.ejhg.5201828. — PMID 17406640. - Laberge A.M., Jomphe M., Houde L., et al.
A «Fille du Roy» Introduced the T14484C Leber Hereditary Optic Neuropathy Mutation in French Canadians (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2005. — Vol. 77, no. 2. — P. 313—317. — doi:10.1086/432491. — PMID 15954041. - Hudson G., Carelli V., Horvath R., Zeviani M., Smeets H.J., Chinnery P.F.
X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy (англ.) // Molecular Vision (англ.)русск. : journal. — 2007. — Vol. 13. — P. 2339—2343. — PMID 18199976. - Hudson G., Keers S., Yu Wai Man P., et al.
Identification of an X-Chromosomal Locus and Haplotype Modulating the Phenotype of a Mitochondrial DNA Disorder (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2005. — December (vol. 77, no. 6). — P. 1086—1091. — doi:10.1086/498176. — PMID 16380918. - Chinnery P.F., Andrews R.M., Turnbull D.M., Howell N.N.
Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation? (англ.) // American Journal of Medical Genetics (англ.)русск. : journal. — 2001. — January (vol. 98, no. 3). — P. 235—243. — doi:10.1002/1096-8628(20010122)98:3<235::AID-AJMG1086>3.0.CO;2-O. — PMID 11169561. - Hudson G., Carelli V., Spruijt L., et al.
Clinical Expression of Leber Hereditary Optic Neuropathy Is Affected by the Mitochondrial DNA-Haplogroup Background (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2007. — August (vol. 81, no. 2). — P. 228—233. — doi:10.1086/519394. — PMID 17668373. - Hoegger M.J., Lieven C.J., Levin L.A.
Differential production of superoxide by neuronal mitochondria (англ.) // BMC Neurosci : journal. — 2008. — Vol. 9. — P. 4. — doi:10.1186/1471-2202-9-4. — PMID 18182110. - ↑ 12Qi X., Sun L., Hauswirth W.W., Lewin A.S., Guy J.
Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation (англ.) // JAMA : journal. — 2007. — February (vol. 125, no. 2). — P. 268—272. — doi:10.1001/archopht.125.2.268. — PMID 17296905. - ↑ 12Ghelli A., Porcelli A.M., Zanna C., Martinuzzi A., Carelli V., Rugolo M.
Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids (англ.) // Investigative Ophthalmology & Visual Science (англ.)русск. : journal. — 2008. — February (vol. 49, no. 2). — P. 671—676. — doi:10.1167/iovs.07-0880. — PMID 18235013. - GeneTests LHON search (недоступная ссылка)
- ↑ 12Klopstock, T.; Yu-Wai-Man, P., Dimitriadis, K., Rouleau, J., Heck, S., Bailie, M., Atawan, A., Chattopadhyay, S., Schubert, M., Garip, A., Kernt, M., Petraki, D., Rummey, C., Leinonen, M., Metz, G., Griffiths, P. G., Meier, T., Chinnery, P. F.
A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy (англ.) // Brain (англ.)русск. : journal. — Oxford University Press, 2011. — 25 July (vol. 134, no. 9). — P. 2677. — doi:10.1093/brain/awr170. - ↑ 12Shrader W. D., Amagata A., Barnes A., Enns G. M., Hinman A., Jankowski O., Kheifets V., Komatsuzaki R., Lee E., Mollard P., Murase K., Sadun A. A., Thoolen M., Wesson K., Miller G.
α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. (англ.) // Bioorganic & medicinal chemistry letters. — 2011. — Vol. 21, no. 12. — P. 3693—3698. — doi:10.1016/j.bmcl.2011.04.085. — PMID 21600768. [] - Sadun A.A., Salomao S.R., Berezovsky A., et al.
SUBCLINICAL CARRIERS AND CONVERSIONS IN LEBER HEREDITARY OPTIC NEUROPATHY: A PROSPECTIVE PSYCHOPHYSICAL STUDY (англ.) // Trans Am Ophthalmol Soc : journal. — 2006. — Vol. 104. — P. 51—61. — PMID 17471325. - Carelli V., Ross-Cisneros F.N., Sadun A.A.
Mitochondrial dysfunction as a cause of optic neuropathies (англ.) // Prog Retin Eye Res : journal. — 2004. — January (vol. 23, no. 1). — P. 53—89. — doi:10.1016/j.preteyeres.2003.10.003. — PMID 14766317. - Katz, Jason; Patel, Chetan.
Parkland Manual of Inpatient Medicine (неопр.). — Dallas, TX: FA Davis, 2006. — С. 903. - Clinical Idebenone trial recruiting at Newcastle University UK https://lhon.ncl.ac.uk
- Mashima Y., Kigasawa K., Wakakura M., Oguchi Y.
Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy? (англ.) // J Neuroophthalmol : journal. — 2000. — September (vol. 20, no. 3). — P. 166—170. — doi:10.1097/00041327-200020030-00006. — PMID 11001192. - Архивированная копия (неопр.)
(недоступная ссылка). Дата обращения 5 июня 2011. Архивировано 4 сентября 2011 года. Sadun,A et al. «EPI-743 alters the natural history of progression of Leber hereditary optic neuropathy». AOS meeting. May 2011] - Newman N.J., Biousse V., David R., et al.
Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite (англ.) // American Journal of Ophthalmology (англ.)русск. : journal. — 2005. — September (vol. 140, no. 3). — P. 407—415. — doi:10.1016/j.ajo.2005.03.058. — PMID 16083844. - Haroon M.F., Fatima A., Scholer S., et al.
Minocycline, a possible neuroprotective agent in Leber’s hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11778 mutation (англ.) // Neurobiology of Disease (англ.)русск. : journal. — 2007. — Vol. 28, no. 3. — P. 237—250. — doi:10.1016/j.nbd.2007.07.021. — PMID 17822909. - Clinical Curcurmin trial recruiting at ClinicalTrials.nlm.nih.gov Архивировано 13 февраля 2009 года.
- Wisconsin near infrared trial Архивировано 15 мая 2008 года.
- Craven L., Tuppen H.A., Greggains G.D., Harbottle S.J., Murphy J.L., Cree L.M., Murdoch A.P., Chinnery P.F., Taylor R.W., Lightowlers R.N., Herbert M., Turnbull D.M.
Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease (англ.) // Nature : journal. — 2010. — May (vol. 465, no. 7294). — P. 82—85. — doi:10.1038/nature08958. — PMID 20393463.
Симптомы наследственной оптической нейропатии Лебера:
Клинически — острое начало потери зрения, сначала в одном глазу, а затем через промежуток времени от нескольких недель до нескольких месяцев — в другом. Начинается обычно в юности, но были сообщения о начальном возрасте в диапазоне 7-75 лет. Начальный возраст немного выше у женщин, (диапазон 19-55 лет: в среднем 31,3 лет), чем мужчин (диапазон 15-53 лет: в среднем 24,3 года). Соотношение между мужчинами и женщинами колеблется в зависимости от мутаций: 3: 1 для 3460 G>A, 6: 1 для 11 778 G>А и 8: 1 для 14484 T>C.
Это, как правило, развивается в очень тяжелую атрофию зрительного нерва и постоянное снижение остроты зрения, отражаясь на оба глаза одновременно (25 % случаев) или последовательно (75 % случаев) со средним интервалом 8 недель. Лишь в редких случаях только один глаз может пострадать. В острой стадии, длящейся несколько недель, пораженный глаз демонстрирует появление отёка слоя нервных волокон, особенно в дугообразных пучках и увеличенных или телеангиэктатических и извилистых околососковых сосудов (микроангиопатии). Главные особенности видны при офтальмоскопии, перед или после потери зрения. Дефекты зрачка также могут быть видны в острой стадии. Анализ показывает снижение остроты зрения, потерю цветового зрения и тифлоцентральную скотому при тестировании поля зрения.
«LHON Плюс» — имя, данное редким случаям болезни глаз при наличии других условий. Симптомы этой высшей формы заболевания включают потерю способности мозга контролировать движения мышц, и сердечную аритмию.Во многих случаях LHON Плюс был сопоставим с рассеянным склерозом из-за недостатка мышечного контроля.
Лечение наследственной оптической нейропатии Лебера:
Без знания истории семьи LHON диагноз, как правило, требует нервно-офтальмологической оценки и анализа крови для митохондриальной оценки ДНК. Здесь важно исключить влияние других возможных причин потери зрения и важных, связанных синдромов, таких как сердечная электрическая проводимость системных аномалий. Прогноз для пострадавших, остающихся неизлечимыми, почти всегда означает продолжение существенного в обоих глазах. Регулярные проверки остроты зрения и проверки периметрии рекомендуется для дальнейших шагов пострадавших лиц. Существует прекрасная терапия для некоторых случаев этого заболевания, особенно для раннего начала болезни. Кроме того, экспериментальные протоколы лечения продолжаются. Должно быть предложено генетическое консультирование. Здоровье и образ жизни должны быть пересмотрены, особенно в свете токсичных и пищевых теорий экспрессии генов. Зрячие помощники и восстановительные работы должны быть использованы для оказания помощи в сохранении рабочих мест.
Для тех, кто являются носителями мутации LHON, доклинические маркеры могут быть использованы для мониторинга прогресса. Например фотография дна может контролировать отёк слоя нервных волокон. Оптическая когерентная томография может быть использована для более детального изучения толщины слоя нервных волокон сетчатки. Тестирование красно-зеленого цветоощущения может обнаружить потери. Контрастная чувствительность может быть уменьшена. Может быть выявлена ненормальная электроретинограмма или зрительный вызванный потенциал. Нейрон-энолаза и маркеры аксонов тяжелой цепи нейрофиламентов крови может предсказать статус преобразования для пострадавших.
Цианокобаламин (форма витамина В12) следует избегать, поскольку это может привести к слепоте у больных болезнью Лебера.
Как правило рекомендуется избегать токсинов зрительного нерва, особенно табак и алкоголь. Некоторые отпускаемые по рецепту лекарства, как известно, несут потенциальный риск, так что ко всем препаратам следует относиться с подозрением и проверить перед использованием степень риска. Этамбутол, в частности, был причастен как импульс к потери зрения у носителей LHON. В самом деле, токсичные и пищевые оптические нейропатии могут иметь пересекающиеся с LHON симптомы, митохондриальные механизмы болезни и управления. Следует отметить, когда пациент в результате LHON или токсической/ пищевой оптической невропатии перенес , усложнюющий процесс болезни, нитропруссид (торговое название: Nipride) не должны использоваться в связи с повышенным риском ишемии зрительного нерва, как следствие реакции на этот антигипертензивный препарат.
Идебенон в небольшом плацебо-контролируемом исследовании, был показан примерно половине пациентов для достижения умеренной пользы. Лучшие результаты были у людей, которые были в начале заболевания.
α-Токотриенол-quinone, метаболит витамина Е, имел в небольших открытых исследованиях некоторый успех в обращении вспять начавшейся потери зрения.
Есть различные подходы к лечению, которые проходили предварительные испытания или предложения, ни один еще не привел убедительных доказательств полезности и безопасности для лечения или профилактики том числе: бримонидин, Миноциклин, куркумин, глутатион, фототерапия инфракрасного излучения, и методы вирусного вектора.
«Экстракорпоральное оплодотворение в третьем лице» является доказательством концепции исследовательских методов для предотвращения митохондриальной болезни в развитии человеческого плода. До сих пор, были произведены жизнеспособные макаки. Но препятствия этического и познавательного характера останавливают использование этого метода на людях.
Митохондриальное нейродегенеративное заболевание с поражением зрительного нерва, часто характеризуется внезапной потерей зрения.
Распространенность
этого заболевания точно неизвестна, но оценивается в 2-4 случая на 100 000 населения.
НОНЛ возникает в результате мутации в митохондриальной ДНК (мтДНК). Доказано, что триггерными механизмами для заболевания могут послужить стресс, курение, алкоголь, токсины, вирусы, прием некоторых лекарственных средств.
Клиника.
Заболевание проявляется внезапной, безболезненной, острой/подострой потерей центрального зрения, обычно в возрасте от 18 до 30 лет.
При НОНЛ поражаются или оба глаза одновременно или последовательно с интервалом в несколько недель или месяцев после первого. Чаще потеря зрения происходит подостро в течение нескольких недель, затем состояние стабилизируется. Однако у многих пациентов в течение нескольких лет продолжаются расширяться размеры центральной скотомы, приводя к глубокой слепоте.
На ранних этапах поражения зрения могут наблюдаться нарушения цветового восприятия красного и зелёного и контрастности.
Могут присутствовать также и другие неврологические симптомы. Эти нарушения известны как «Лебера плюс», и включают двигательные расстройства, дистонию, постуральный тремор и мозжечковую атаксию.
Диагноз
ставится на основании офтальмоскопической экспертизы. Признаки НОНЛ при офтальмоскопии включают отек диска зрительного нерва, извитые сосуды, перипапиллярные телеангиоэктазии, микроангиопатии и центральные скотомы при визуальном тестировании полей зрения.
Оптическая когерентная томография (ОКТ) помогает подтвердить отек слоя нервных волокон сетчатки. Еще до снижения зрения у пациентов- носителей мутации удается выявить нарушение цветового восприятия «красный-зеленый», а также сниженные или пограничные показатели электроретинограммы и зрительных вызванных потенциалов.
При дифференциальной диагностике, в первую очередь, следует исключить рассеянный склероз, при котором распространённым признаком является неврит зрительного нерва. Также необходимо исключить другие генетические оптические невропатии, такие как синдром Вольфрама и классические аутосомно-доминантные типы атрофии зрительного нерва.
Лечение.
Не существует специфического лечения для НОНЛ. Основной поддерживающей терапии являются препараты для слабовидящих. Несколько веществ показали положительные результаты в восстановлении зрения. Синтетический аналог коэнзима Q10 — идебенон улучшил зрение после года применения.
В настоящее время проходит испытание третье поколение хинонов, и также есть сообщения о положительном эффекте. Очень важно, чтобы пациент исключил употребление алкоголя, табака и прием некоторых антибиотиков, которые также влияют на митохондриальное окислительное фосфорилирование.
Прогноз
заболевания зависит от возраста появления симптомов. У молодых прогноз более благоприятный. При некоторых мутациях описано спонтанное частичное восстановление зрения через 1-2 года после дебюта заболевания. У 30-50% мужчин и 80-90% женщин-носителей мутации, слепота не наступает. Полная слепота развивается крайне редко.
Link to Orphanet
| Leber optic atrophy |
Справка
На сегодняшний день в России редкими предлагается считать заболевания с распространенностью не более 10 случаев на 100 000 человек.
В список орфанных болезней в России внесено 215 заболеваний. (список Минздрава от 7.05.2014)
Также хотим обратить Ваше внимание на следующие документы, добавленные нами в соответствующие разделы нашей Энциклопедии.
Наследственная Оптическая Нейропатия Лебера (NOHL или LHON), также известная как Атрофия Зрительного Нерва Лебера (LOA), названа в честь доктора Теодора Лебера, который описал её в 1871 году, как характерную картину внезапной потери зрения у молодых людей, в с семейном анамнезе которых наблюдалась потеря зрения. Это самая распространенная наследственная оптическая невропатия, её причиной является митохондриальная мутация и её распространенность очень низкая, их в большинстве географических районов она неизвестна. На северо-востоке Англии и Финляндии заболевание затрагивает 1 человека из каждых 31 000 или 50 000 соответственно.
Это обычно наблюдается у молодых мужчин в возрасте от 18 до 35 лет, хотя это может также повлиять на детей младшего возраста и взрослых старше 35 лет. У женщин оно встречается гораздо реже.
Как правило, оно вызывает серьезную потерю зрения в обоих глазах. В большинстве случаев, процесс начинает в одном глазу и через несколько недель или месяцев переходит на второй глаз.
Почему оно производится? Что такое наследственность митохондрий?
Хотя подавляющее большинство нашего генетического материала (ДНК) расположено в ядре клеток, его небольшая часть находится в митохондриях (митохондриальная ДНК или мтДНК).
Гены ядра наследуются от обоих биологических родителей. Однако, ДНК митохондрии наследуются только от матери. Это означает, что мужчина с мутацией в митохондриальной ДНК не передаст его ни одному из своих биологических потомков, тогда как женщина с мутацией в митохондриальной ДНК передаст её всем своим биологическим потомкам.
90-95% случаев LHON связаны с одной из трех специфических мутаций мтДНК. Однако значительная часть людей с этими мутациями никогда не развивают симптомы заболевания. В частности, только 10% женщин и 50% мужчин, несущих любую из этих мутаций, имеют оптическую нейропатию . Это подразумевает, что другие факторы, генетические (митохондриальные или ядерные) и / или факторы окружающей среды должны действовать так, чтобы произошла потеря зрения. Так, например, воздействие табака или большого количества алкоголя у людей, несущих эти мутации, увеличивает риск развития заболевания.
Симптомы Оптической Нейропатии Лебера
Как правило, носители LHON не имеют до тех пор, пока они не теряют значительно быстро зрение в одном глазу и через несколько недель или месяцев также в другом глазу. Зрение продолжает ухудшаться в течение нескольких недель, пока оно не станет значительным, и в большинстве случаев центральное видение (необходимое для чтения, вождения и распознавания лиц) серьезно страдает в обоих глазах, хотя некоторые люди испытывают определенное восстановление зрения через некоторое время. Эта постоянная потеря зрения является следствием смерти клеток зрительного нерва, ответственных за передачу изображений из глаз в головной мозг.
Хотя потеря зрения является единственным симптомом у большинства пациентов с LHON, в некоторых случаях из них были описаны нарушения сердечного ритма, а также неврологические изменения (такие как постуральный тремор или другие нарушения движения).
Как оно диагностируется? Существует лечение этого заболевания?
Это заболевание сложно диагностировать по нескольким причинам. Во-первых, это наследственное заболевание, но оно не проявляется у всех людей, имеющих мутации. По этой причине он может появиться без учета семейного анамнеза болезни.
Чтобы диагностировать его, обычно необходимо провести тщательную нейрофтальмологическую оценку и анализ крови для исследования мтДНК.
В анализе глазного дна сначала наблюдается микроангиопатия и отек слоя перипапиллярного нервного волокна, который прогрессирует до оптической атрофии.
В настоящее время прогноз обычно представляет собой постоянную серьезную потерю зрения. Изучается несколько способов лечения этого заболевания, и в ICR проводится исследование болезни, которая находится на этапе отбора.
Амавроз Лебера – генетическое офтальмологическое заболевание, приводящее к значительному ухудшению зрительной функции (вплоть до ее полной утраты). В его основе лежит необратимое поражение светочувствительных клеток сетчатой оболочки глазного яблока (колбочек и палочек).
Амавроз Лебера относится к редким наследственным патологиям: он встречается с частотой 3 случаев на каждые 100 000 новорожденных. Примерно каждый 5-й случай врожденной слепоты или слабовидения обусловлен амаврозом Лебера.
Причины и факторы риска
Врожденный амавроз Лебера – это целая группа дистрофий сетчатки, вызванных различными генетическими причинами. Наиболее часто она наследуется по аутосомно-рецессивному типу, то есть для развития патологии ребенок должен получить мутированный ген и от матери, и от отца.
Прогноз при амаврозе Лебера негативный: дети либо необратимо слепы от рождения, либо теряют зрение в течение первых десяти лет жизни.
Существует несколько форм амавроза Лебера, которые наследуются по аутосомно-доминантному типу. В этой ситуации для возникновения дистрофического процесса в сетчатой оболочке глазного яблока достаточно только одного измененного гена.
Признаки и симптомы
Снижение зрительной реакции при рождении является первым признаком заболевания. Часто ребенок тыкает, нажимает и протирает глаза, чтобы стимулировать сетчатку глаза для получения света. Эта деятельность может привести к тому, что глаза станут запавшими или глубоко посаженными (энофтальм).
Другие симптомы могут включать:
- косоглазие;
- нистагм;
- светобоязнь;
- катаракта;
- кератоконус.
Кроме того, у некоторых детей может быть потеря слуха, умственная отсталость и/или задержка развития.
Формы заболевания
В зависимости от того, в каком именно гене произошла мутация, выделяют несколько типов заболевания:
- Мутации в гене CRX, ответственном за развитие у эмбриона фоторецепторов, а также поддержание необходимого их количества в течение жизни. Мутации в нем вызывают позднюю пигментную дегенерацию сетчатки, амавроз Лебера VII типа и палочко-колбочковую дистрофию. Все эти формы заболевания имеют аутосомно-доминантный тип наследования.
- Мутации в гене LCA5, кодирующем синтез белка леберцилина клетками сетчатки. Его мутации приводят к амаврозу Лебера V типа с аутосомно-доминантным типом наследования.
- Мутации в гене RPE Он несет код синтеза белка, специфичного для пигментного эпителия сетчатки и играющего ведущую роль в метаболизме ретинола. Описано более 80 вариантов мутации гена RPE65, которые влекут пигментную дегенерацию сетчатки, амавроз Лебера II типа.
- Мутации в гене LRAT, ответственном за синтез микросомального белка – лецитин-ретинол-ацилтрансферазы (LRAT). Он участвует в процессе обмена ретинола (витамина A) в клетках системы зрения и печени. Мутации гена становятся причиной пигментной ювенильной дегенерации сетчатки, амавроза Лебера XIV типа.
- Мутации в гене CRB1, кодирующем синтез белка, необходимого для нормального развития и функционирования фоторецепторов. В настоящее время известно свыше 140 различных мутаций данного гена, которые приводят к пигментной дегенерации сетчатки XII типа, амаврозу Лебера VIII типа.
Примерно каждый 5-й случай врожденной слепоты или слабовидения обусловлен амаврозом Лебера.
Генетика
Наследственная оптическая нейропатия Лебера имеет митохондриальный шаблон наследования.
Наследственная оптическая невропатия Лебера является состоянием, связанным с изменениями в митохондриальной ДНК . Хотя большинство ДНК упаковано в хромосомах внутри ядра, митохондрии имеют особый митохондриальный геном, состоящий из мтДНК.
Мутации генов МТ-ND1 , МТ-ND4 , МТ-ND4L и MT-Nd6 вызывают наследственную оптическую невропатию Лебера.[10] Эти гены кодируют мембранную часть белка НАДН-дегидрогеназы, участвующего в нормальной митохондриальной функции окислительного фосфорилирования . Окислительное фосфорилирование использует серию из четырех крупных мультиферментных комплексов, которые все встроены во внутреннюю мембрану митохондрий для преобразования кислорода и моносахаридов в энергию. Мутации в любом из генов нарушают этот процесс, вызывая различные синдромы в зависимости от типа мутации и других факторов. Остается неясным, как эти генетические изменения приводят к гибели клеток в зрительном нерве и к другим специфическим симптомам наследственной оптической нейропатии Лебера.
Диагностика
Диагностика амавроза Лебера начинается с проведения офтальмологического осмотра. Затем применяют следующие методики:
- зрительные вызванные потенциалы (ЗВП);
- электроретинография;
- электроокулография.
Наибольшее диагностическое значение имеет электроретинография (ЭРГ). Метод позволяет зарегистрировать биоэлектрическую активность нейронов сетчатки. При амаврозе Лебера ЭРГ не регистрируется. При врожденной слепоте, вызванной другими причинами (сифилисом, краснухой, атрофией зрительного нерва), ЭРГ показывает нормальную или субнормальную картину.