Lissencephaly - main symptoms:
- Convulsions
- Epileptic seizures
- Delayed speech development
- Small head
- General swelling
- Lethargy
- Skull deformation
- Stunted growth in a child
- Deformation of the facial structure
- Disorders of the gastrointestinal tract
- Kidney dysfunction
- Cardiac dysfunction
- Mental development disorder
- Muscle hypertonicity
- Retarded physical development
- Muscle dystrophy
- Sloping forehead
- Retinal detachment
- Large distance between eyes
- Increased reflexes
What is lissencephaly
Lissencephaly or smooth brain is a developmental anomaly characterized by complete or partial smoothing of the gyri or sulci of the cerebral hemispheres. The pathology is considered quite rare, as it occurs in 11 children per 1 million births.
The main reason for the formation of any type of disease is insufficient migration of germinal nerve cells or neuroblasts from the primary neural tube. In addition, there are several other predisposing factors.
The severity of clinical manifestations depends on the course of the disease. The most common symptoms include seizures, mental retardation, decreased muscle tone, and impaired swallowing.
Diagnosis is based on information obtained during instrumental examinations. It is worth noting that the correct diagnosis can be made even at the stage of intrauterine development of the fetus.
The peculiarity of the disease is that today there is no specific treatment. Therapy is aimed at relieving some symptoms and maintaining the patient’s normal condition.
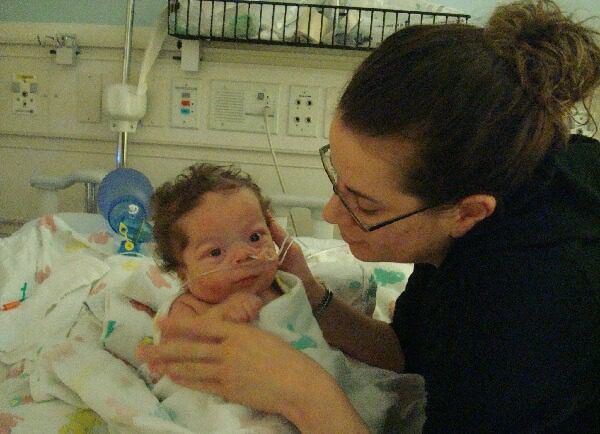
A rare brain defect has become the cause of a terrible disease. Only a complicated operation will save Ksyusha
One-year-old Ksyusha is almost no longer afraid of doctors and smiles at everyone during medical examinations. The baby spent most of her life in the hospital with her mother, practically never being at home.
The baby has structural focal epilepsy, drug-resistant course. This means that no medications can completely stop epileptic seizures.
The girl needs complex brain surgery as soon as possible.
“Ksyusha is our second daughter, a welcome child. We prepared for her appearance in the most thorough manner. When we finally found out that there would soon be a new addition to our family, happiness knew no bounds. The pregnancy proceeded without complications; at that time I was still on maternity leave with my eldest. We spent time together, walked a lot, relaxed and looked forward to the arrival of the baby. We dreamed of how the two sisters would finally meet and enjoy a carefree childhood together,” recalls the mother.
All tests and ultrasound were normal. The girl was born at term with high Apgar scores. On the third day, mother and newborn were discharged home, and pleasant chores began. The parents did not notice anything unusual. Until four months, the baby developed according to the norms.
By two months she had learned to hold her head up, by three months she was already rolling over, was interested in toys, often smiled and walked. After some time, the parents noticed that their daughter began to have difficulty sleeping during the day. The baby was examined and a neurosonogram was performed, but nothing suspicious was found.
The doctors assured the parents that everything was fine with the child, the girl was active, would sit up early and walk early.
But after another month everything changed. Ksyusha began to have convulsions. The parents immediately went to the hospital in the hope of quickly receiving treatment recommendations and returning home. After all, just recently everyone said that Ksyusha was completely healthy. But plans have changed.
A series of examinations followed in different clinics. After some consultations, the mother and baby went to others, but there was still no diagnosis. Ksyusha was not home for weeks and did not see her older sister and dad.
Mom spent sleepless nights near the crib, counting every convulsive spasm.
Six months later the final diagnosis was made. It turned out that Ksyusha had a congenital brain defect (polymicrogyria of the right frontal region), which led to epilepsy. Medicines can temporarily reduce the frequency and intensity of attacks, but not stop them completely.
And this threatens a lag in physical and mental development, as well as a loss of existing skills. Already now Ksyusha is lagging behind her peers. The situation may worsen even further. The only thing that will help the baby is radical surgery - vertical parasagittal hemispherotomy on the right.
“Unfortunately, the capabilities of our equipment cannot keep up with such complex diseases. Doctors' opinions regarding surgical intervention differed. At one of the many consultations, we were advised to look for a clinic with the most modern equipment to determine the source of epilepsy. With the help of relatives and personal savings, we managed to get to a place where they have been treating children like Ksyusha for many years,” said the mother.
After a high-precision examination, which lasted several days in a row, it was possible to detect foci of epilepsy and develop a surgical plan. Treatment is required as soon as possible.
Each new attack that occurs in the right hemisphere negatively affects the healthy left. During the operation, the girl’s right hemisphere will be almost completely “turned off.” Unfortunately, this is the only possible method of radical treatment.
After the operation, the baby will have a long and difficult rehabilitation to restore the physical capabilities of her body. “The sooner this is done, the faster Ksyusha will begin to develop normally and enjoy childhood without pain. There are two techniques for performing this operation. The method that will be used to operate on my daughter in Germany is characterized by less blood loss and trauma. And this will greatly affect the result of the operation and further recovery. Only the bill separates us from the long-awaited deliverance,” Ksyusha’s mother shared.
The cost of the operation is 50,572 euros, or 3,707,433 rubles at the Alfa-Bank rate of 71.31+2 rubles. for 1 euro. The family spent all their savings on expensive pre-operative testing and are unable to pay for emergency surgery. We ask you to help Ksyushenka become healthy and develop like all the other children.
HELP KSUSHA:
Source: https://zen.yandex.ru/media/aleshafond/redchaishii-porok-mozga-stal-prichinoi-strashnoi-bolezni-ksiushu-spaset-tolko-slojnaia-operaciia-5d5bde169f272100ad2c2e1c
Causes of lissencephaly
Lissencephaly is a group of severe brain developmental defects that can be accompanied by conditions such as pachygyria and agyria. In the first case, the presence of only a few grooves and convolutions is noted, and in the second they are completely absent.
Such an anomaly can act as an independent genetic disorder or be part of a symptom complex of other congenital syndromes, for example, Miller-Dieker, Fukuyama or Walker-Warburg.
Geneticists identify several options for inheriting such a disorder:
- autosomal recessive;
- autosomal dominant;
- linked to the X chromosome.
Disturbances in the migration of nerve cells at the stage of formation of the cerebral cortex during intrauterine development of the fetus are caused by:
- viral infections suffered by the expectant mother in the 1st trimester of pregnancy - this includes rubella, cytomegalovirus or herpes virus infection;
- parasitic infestations, in particular toxoplasmosis;
- burdened heredity;
- improper formation of cerebral vessels, and children develop a lack of cerebral circulation.
With some types of mutations that provoke the formation of such a defect, the possibility of developing anomalies in other internal organs and systems cannot be ruled out, which further aggravates the already serious condition of patients.
Brain development abnormalities
Anomalies of brain development are defects consisting of abnormal changes in the anatomical structure of cerebral structures. The severity of neurological symptoms accompanying cerebral anomalies varies significantly. In severe cases, defects are the cause of antenatal fetal death; they account for up to 75% of cases of intrauterine death. In addition, severe cerebral anomalies account for about 40% of newborn deaths. The timing of manifestation of clinical symptoms may vary. In most cases, cerebral abnormalities appear in the first months after the birth of the child. But, since the formation of the brain lasts until the age of 8, a number of defects make their clinical debut after the 1st year of life. In more than half of the cases, cerebral defects are combined with defects of somatic organs: congenital heart defects, renal fusion, polycystic kidney disease, esophageal atresia, etc. Prenatal detection of cerebral anomalies is an urgent task of practical gynecology and obstetrics, and their postnatal diagnosis and treatment are priority issues of modern neurology, neonatology, pediatrics and neurosurgery.
Brain Formation
The construction of the fetal nervous system begins literally from the first week of pregnancy. Already by the 23rd day of gestation, the formation of the neural tube ends, incomplete fusion of the anterior end of which entails serious cerebral anomalies. By approximately the 28th day of pregnancy, the anterior cerebral vesicle is formed, which subsequently divides into 2 lateral ones, which form the basis of the cerebral hemispheres. Next, the cerebral cortex, its convolutions, corpus callosum, basal structures, etc. are formed.
Differentiation of neuroblasts (germinal nerve cells) results in the formation of neurons, which form the gray matter, and glial cells, which make up the white matter. Gray matter is responsible for higher processes of nervous activity. The white matter contains various pathways that connect cerebral structures into a single functioning mechanism. A full-term newborn has the same number of neurons as an adult. But the development of his brain continues, especially intensively in the first 3 months. life. There is an increase in glial cells, branching of neuronal processes and their myelination.
Classification
Based on the location of the mutant gene, the following types of pathology are distinguished:
- Form 1 is a consequence of changes in the PAFAH1B1 gene, which is also known as LIS1 and is located on chromosome 17. Often the surrounding segments are damaged.
- Form 2 – caused by a mutation of the DCX gene, localized on the X chromosome. It is for this reason that the inheritance of this type of disease directly depends on gender. Another type is associated with mutations of the ARX gene, which controls the formation and functioning of the pancreas or reproductive system. This indicates that in the presence of this type of disorder, defects on the part of these organs appear.
- Form 3 – develops against the background of changes in the RELN gene, located on chromosome 17. In this case, the formation of the so-called Norman-Roberts syndrome occurs. This category also includes the TUBA1A gene, which is located on chromosome 12.
Currently, specialists know more than 20 types of lissencephaly, which are divided into several classes:
- 1 – is called classical lissencephaly. It combines the first two of the above forms, as well as types of pathology that develop for unknown genetic reasons. A distinctive feature is the presence of minimal disturbances in the structure of the brain.
- 2 – consists of only one type, caused by changes from ARX. Patients have an absence of the corpus callosum, problems with thermoregulation and other abnormalities.
- 3 – combines those variants that are combined with hypoplasia or complete underdevelopment of the cerebellum, which is most typical for RELN mutations.
- 4 – microlissencephaly. This class received this name due to the fact that in addition to the incorrect formation of the cerebral cortex, pronounced microcephaly occurs.
- 5 – or cobblestone lissencephaly. This includes Fukuyama and Walker-Warburg syndrome.
This classification is generally accepted, but is far from complete, since it does not include less common types of the disease or those that arise for unknown reasons.
According to severity they are distinguished:
- complete argyria;
- diffuse argyria with the presence of several folds on the frontal or occipital pole;
- a mixture of agyria and pachygyria;
- diffuse or mixed pachygyria with normal gyri;
- anterior mixed pachygyria, combined with subcortical heterotopias;
- isolated subcortical heterotopias.
Polymicrogyria: causes, symptoms and consequences
Typically, during normal brain development, a series of folds or turns are formed that serve to make the surface of the brain take up less space and fit inside the skull. Like any other organ, the brain also suffers from developmental defects. An example of this is polymicrogia, which affects the morphology of cortical folds. .
It is a disease that causes severe neurological symptoms due to malformations that occur during fetal development. Let's take a quick look at what learning problems we find in polymicroria, what causes it, and what can be done about those people who suffer from it.
What is polymicrogia?
The etymology of the word poly (multiple) micro (small) -gyria (folds) clearly indicates this: it is a defect in morphology in which a larger number of small folds are observed.
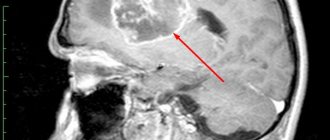
This genetic defect in the cortex can be clearly detected using x-ray diagnostic tests such as magnetic resonance imaging (MRI).
In these images you can see how the cortex becomes thicker and more intense, indicating higher density, and how the folds appear superficial, unlike normal brain.
There are different forms of polymicrogyria, some of which are more serious. When it affects only one part of the brain, it is called unilateral. However, it is called bilateral when both hemispheres are affected.
The nomenclature refers not only to symmetry, but also to how many areas of the brain are affected.
Thus, a brain that has only one affected area will suffer from focal polymicrogyria compared to generalized polymicrogyria where it affects almost all or all of the cortex.
Polymicrogyria patterns favor some lobes over others . The most commonly affected lobes are the frontal lobes (70%), followed by the parietal lobes (63%), and then the temporal lobe (18%). Only a small proportion (7%) affected the occipital lobes. In addition, it seems to tend to distort the lateral fissures that separate the frontal and parietal lobes from the temporal lobe (60%) more often.
- Article on the topic: “Lobes of the brain and its various functions”
Signs and symptoms
Although focal and unilateral forms are usually no more problematic than drug-controlled seizures, when they are bilateral, the severity of symptoms increases. Some symptoms include:
- epilepsy
- Developmental delay
- strabismus
- Problems with speech and swallowing
- Muscle weakness or paralysis
The global impacts of polymicrogyria have profound mental retardation, severe movement problems, cerebral palsy, and seizures cannot be controlled with medications.
For this reason, while milder forms of polymicrogyira allow a very long life expectancy, people who are born with the severe disease die when they are very young due to complications of the disease.
Often polymicrogia does not occur separately or purely, but together with other syndromes, such as:
- Variants of Adams-Oliver syndrome
- Arima syndrome
- Galloway-Mowatt syndrome
- Delleman syndrome
- Zellweger syndrome
- Fukuyama muscular dystrophy
causes
In most cases the cause is unknown. There is a percentage of cases where the mother suffers from intrauterine infection during pregnancy. Some viruses that are associated with the development of polymicrogyria are cytomegalovirus, toxoplasmosis , syphilis, and varicella zoster virus.
Hereditary causes include both chromosomal syndromes, that is, they affect several genes at the same time, like single-gene syndromes. There are many genetic disorders that change the way the brain is formed. Because of this, many genetic diseases are accompanied by polymicrogyria, among other manifestations.
The GPR56 gene has been identified as one of the main causes of polymicrogyria in its global and bilateral versions.
One study found that all patients studied had some modification of this gene, which led to damage to the central nervous system.
It is known that this gene is very involved in the formation and development of the fetal cerebral cortex during pregnancy.
Therefore, it is important that parents who suffer from or are at risk of polymicrogyria are informed of the hypothetical risk of passing on their condition to their child and, through genetic testing, find out what the real likelihood of their occurrence is before pregnancy.
Patient management after diagnosis
Once diagnosed through imaging, it will be necessary to conduct a full assessment in the areas affected by polymicrogyria . Pediatricians, neurologists, physical therapists and occupational therapists should intervene to assess the risk of developmental delay, mental retardation or even cerebral palsy. In this sense, special educational needs will be identified so that the patient can learn at the speed allowed by his disease. Speech will be assessed in those with lateral clefts, vision and hearing.
Symptoms will be treated with physical therapy, pharmacological intervention with antiepileptic drugs, orthopedic medications and surgery for patients suffering from muscle stiffness. When speech problems arise, speech therapy and professional intervention are provided.
Ultimately, parent education is the cornerstone of symptom management. They will need to be educated on how seizures occur and what to do when they occur. Additionally, supportive measures can be used to prevent joint problems or bedsores from occurring due to sitting in the same position for too long.
Symptoms of lissencephaly
The first clinical manifestations begin to appear at 3-5 months of the baby’s life, extremely rarely this occurs by 8-9 months. In this case, the symptoms will be completely dictated by the type of disease. For example, classic lissencephaly is represented by:
- small head size;
- pronounced retardation in growth, as well as speech, mental and motor development;
- convulsive seizures - they can be expressed from the first days of life or manifest after 1.5 years;
- lethargy of the child;
- sloping forehead;
- large distance between the eyes;
- muscle hypertonicity with increased reflexes.
With Miller-Dickens syndrome, symptoms include:
- changing the shape of the face;
- increased distance between paired internal organs;
- slow growth;
- dysfunction of the gastrointestinal tract, kidneys and heart.
In the case of Norman-Roberts syndrome, the symptoms will be as follows:
- epilepsy attacks;
- severe swelling of tissues;
- mental retardation;
- small head;
- deformation of the skull.
Walker-Warburg syndrome is represented by the following pathologies:
- cataract;
- retinal detachment;
- dropsy of the brain;
- skeletal and muscle abnormalities;
- problems with mental development.
Fukuyama syndrome is characterized by the following symptoms:
- convulsions;
- progressive muscular dystrophy;
- delay in the pace of development;
- severe dementia;
- hydrocephalus;
- atrophy of the optic nerves.
It is impossible to accurately determine how the disease progresses, because each patient has an individual symptom complex.
Types of brain development abnormalities
Anencephaly is the absence of a brain and acrania (absence of skull bones). The place of the brain is occupied by connective tissue growths and cystic cavities. May be covered with skin or bare. The pathology is incompatible with life.
Encephalocele is a prolapse of cerebral tissues and membranes through a defect in the bones of the skull, caused by its non-fusion. As a rule, it is formed along the midline, but it can also be asymmetrical. A small encephalocele may mimic a cephalohematoma. In such cases, skull radiography helps determine the diagnosis. The prognosis depends on the size and content of the encephalocele. If the protrusion is small in size and there is ectopic nerve tissue in its cavity, surgical removal of the encephalocele is effective.
Microcephaly is a decrease in the volume and weight of the brain due to its underdevelopment. Occurs with a frequency of 1 case per 5 thousand newborns. Accompanied by a reduced head circumference and a disproportionate ratio of the facial/cranial skull with a predominance of the former. Microcephaly accounts for about 11% of all cases of mental retardation. With severe microcephaly, idiocy is possible. Often there is not only mental retardation, but also a lag in physical development.
Macrocephaly is an increase in brain volume and mass. Much less common than microcephaly. Macrocephaly is usually combined with disorders of brain architecture and focal heterotopia of the white matter. The main clinical manifestation is mental retardation. Convulsive syndrome may occur. Partial macrocephaly occurs with enlargement of only one of the hemispheres. As a rule, it is accompanied by asymmetry of the cerebral part of the skull.
Cystic cerebral dysplasia is characterized by multiple cystic cavities in the brain, usually connected to the ventricular system. Cysts can vary in size. Sometimes they are localized only in one hemisphere. Multiple brain cysts manifest as epilepsy that is resistant to anticonvulsant therapy. Single cysts, depending on their size, may have a subclinical course or be accompanied by intracranial hypertension; their gradual resorption is often noted.
Holoprosencephaly is the absence of separation of the hemispheres, as a result of which they are represented by a single hemisphere. The lateral ventricles are formed into a single cavity. Accompanied by severe dysplasia of the facial skull and somatic defects. Stillbirth or death occurs on the first day.
Agyria (smooth brain, lissencephaly) is underdevelopment of the gyri and severe violation of the architectonics of the cortex. Clinically manifested by a severe disorder of mental and motor development, paresis and various forms of seizures (including West syndrome and Lennox-Gastaut syndrome). Usually ends in death in the first year of life.
Pachygyria is an enlargement of the main gyri in the absence of tertiary and secondary ones. Accompanied by shortening and straightening of the grooves, a violation of the architectonics of the cerebral cortex.
Micropolygyria - the surface of the cerebral cortex is represented by many small convolutions. The bark has up to 4 layers, whereas the normal bark has 6 layers. May be local or diffuse. The latter, polymicrogyria, is characterized by plegia of facial, masticatory and pharyngeal muscles, epilepsy with onset in the 1st year of life, and oligophrenia.
Diagnostics
Changes in the structure of the brain can only be detected using instrumental procedures. However, in order to make an accurate diagnosis, it is necessary to carry out a whole range of diagnostic measures.
First of all, the neurologist needs to:
- study family medical history;
- collect and analyze the patient’s life history: in addition to general information, this includes information regarding the course of pregnancy;
- conduct a thorough physical examination;
- measure the baby's head circumference;
- check reflexes;
- interview the patient’s parents in detail to draw up a complete symptomatic picture.
Instrumental diagnostics combines the most informative procedures:
- CT;
- MRI;
- EEG.
In addition, genetic tests may be required, as well as consultations with a geneticist and neurosurgeon.
As for prenatal diagnosis, pathology can be detected during a routine ultrasound at 22-28 weeks of fetal development. If you suspect the presence of such a deviation, it is necessary to study the child’s chromosomes. In cases where the diagnosis is confirmed, the question of termination of pregnancy is raised.
Treatment of lissencephaly
Specific treatment tactics have not currently been developed. To eliminate symptoms and maintain the patient’s normal condition, the following medications are used:
- anticonvulsants;
- nootropics;
- vitamin complexes;
- Carnitine;
- Glycine;
- Cerebrolysin;
- Actovegin.
Sometimes surgery is performed to perform brain shunting and stem cell transplantation. Operations are performed quite rarely, since they provide short-term improvement and cause a large number of complications.
If there are other pathologies, they are corrected according to individual indications.
Prevention and prognosis
It is not possible to prevent the development of the disease, since it is associated with gene disorders. Nevertheless, the main preventive measures are:
- consultation with a geneticist before conceiving a child - indicated for couples in whose families there is a similar deviation;
- control over the adequate course of pregnancy;
- regular routine ultrasound examinations in each trimester of gestation.
Lissencephaly of any form has an extremely unfavorable prognosis - in the vast majority of cases, patients die in early childhood. Some patients live their lives on the verge of a vegetative state. It is extremely rare for people with this diagnosis to survive into adolescence or adulthood, and they continue to have underdevelopment of the central nervous system for the rest of their lives. The main causes of death are complications associated with other developmental defects, aspiration pneumonia and cardiovascular accidents.
If you think you have lissencephaly
and symptoms characteristic of this disease, then you.
Source
Did you like the article? Share with friends on social networks: