Исследования
Некоторые из передовых стратегий включают в себя: введение гена дистрофина; изменение способов интерпретации клетками генных инструкций дистрофина; исправление самого гена; управление другими белками организма для компенсации отсутствия/нехватки дистрофина; совершенствование стероидных препаратов, и использование стволовых клеток для восстановления поврежденных мышц1.
Другие исследования сосредоточены на лечении сердечной недостаточности, связанной с недостатком дистрофина.
Повреждения гена дистрофина приводят к мышечной дистрофии Дюшенна (МДД), таким же образом, как и при менее тяжелой форме — мышечной дистрофии Беккера (МДБ). Множество стратегий лечения, опробованных при МДД, применимы для мышечной дистрофии Беккера.
Чтобы узнать больше информации об исследованиях, посвященных МДБ, предлагаем ознакомиться с данными видеоматериалами: клинические исследования мышечной дистрофии на животных моделях и МДБ: От целей до клинических испытаний.
Кардиологическая поддержка
Исследователи преследуют несколько стратегий для поддержки и улучшения сердечной функции при МДБ и МДД. Они в основном тестируют существующие препараты на предмет возможной пользы для пораженного при МДД и МДБ сердца и проводят исследования с целью понять и найти новые подходы в лечении сердца при таких заболеваниях.
В 2009 году исследователи выяснили, что мутации в гене дистрофине, вызывающие кардимиопатию при МДБ, могут не поражать регионы белка, отвечающие за потерю скелетных мышц. Исследования могут позволить лучше прогнозировать появление кардиомиопатии при МДБ и применять кардиопротекторную терапию на более ранних этапах. Также они дают исследователям представление о том, какие части белка необходимо сохранить при рассмотрении укороченных молекул дистрофина в качестве терапевтических стратегий.
Обнаружено, что лекарственное средство силденафил (виагра) оказывает кардиопротекторный эффект на мышей с заболеванием, подобным МДД, как на ранней, так и на поздней стадии. Силденафил, используемый для лечения эректильной дисфункции, принадлежит к классу ингибиторов фосфодиэстеразы-5 (ФДЭ5). Он расслабляет гладкую мускулатуру, выстилающую кровеносные сосуды, увеличивая приток крови к мышцам и сердцу. Кардиальные эффекты силденафила у подростков и мужчин с МДД изучаются.
Лабораторные исследования показали, что экспериментальное соединение, предназначенное для герметизации клеточных мембран — р 188, улучшает работу сердца у собак с дефицитом дистрофина.
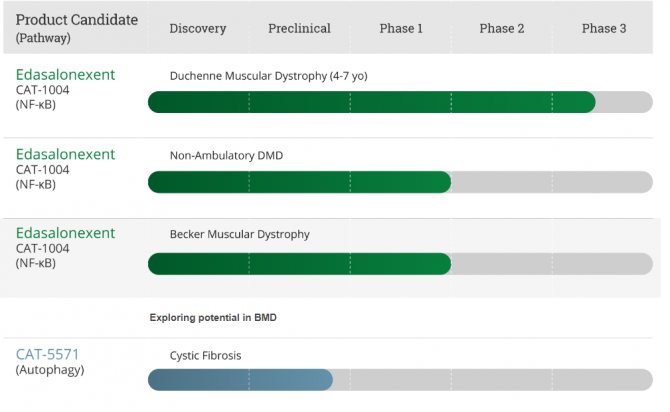
В 2011 году исследователи при поддержке MDA обнаружили, что ингибирование действия белка NF-kappa B улучшает сердечную функцию у мышей с тяжелой болезнью, подобной МДД. Фармацевтическая компания Catabasis, предоставляет на своем официальном сайте инфографический материал о препарате Edasalonexent (CAT-1004) — ингибиторе белка NF-kappa B.
График исследований Сatabasis
Согласно информации, представленной в данном графике, в настоящее время изучается потенциал препарата Edasalonexent (CAT-1004) при лечении МДБ2.
Лечение прогрессирующей мышечной дистрофии Беккера
- Симптоматическая терапия.
- Дозированная ЛФК.
- Глюкокортикостероиды.
- Хирургическое лечение контрактур.
Лечение назначается только после подтверждения диагноза врачом-специалистом.
Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.
- Преднизолон (системный глюкокортикостероид). Режим дозирования: внутрь, в дозе 20-80 мг/сут. на 3-4 приема. Поддерживающая доза 5-10 мг/сут.
- Метиландростендиол (анаболическое стероидное средство). Режим дозирования: сублингвально, взрослым в дозе 0,025–0,050 г/сут., детям в дозе 1,0–1,5 мг/кг/сут. на 3-4 приема.
- АТФ (средство, улучшающее трофику мышц). Режим дозирования: в/м, в дозе 1 мл 1% раствора 1–2 раза в сутки. На курс лечения — 30–40 инъекций. Повторный курс — через 1–2 мес.
Генное редактирование — CRISPR/Cas9
Метод CRISPR/Cas9 основан на естественной системе защиты бактерий от вирусной инфекции (аналог иммунной системы). Когда бактерия обнаруживает наличие чужеродной (в данном случае вирусной) ДНК, CRISPR белок захватывает часть вирусной ДНК и вставляет этот фрагмент в собственный геном бактерии. Затем бактерии используют «иммунизирующий» фрагмент вирусной ДНК для производства «антител», которые распознают и защищают от будущих вирусных атак3.
Бактериальные «антитела» включает в себя два типа коротких РНК,которые образуют комплекс с белком Cas9. Cas9 — это нуклеаза (тип фермента, способный разрезать ДНК). Когда соответствующая вирусу последовательность нуклеотидов (гидовая РНК) обнаруживает цель в геноме вируса, Cas9 вырезает вирусную ДНК, блокирует вирус и препятствует его репликации.
Как CRISPR/Cas9 может быть использован в лечении мышечной дистрофии?
Система CRISPR/Cas9 может быть задействована для изменения или исправления мутаций в клетках пациентов. Исследователи работают над тем, чтобы найти лучший способ лечения различных типов мышечной дистрофии (и других генетических заболеваний) с помощью данного метода.
Первые клинические испытания CRISPR/Cas9 с участием человека для лечения таких заболеваний как рак или муковисцидоз, находятся в процессе разработки. В данных испытаниях клетки пациента изолируются, обрабатываются для исправления генетической мутации, а затем вводятся обратно пациенту для борьбы с болезнью.
Для мышечной дистрофии вирусная система доставки могла бы обеспечить клетки пациента инструкциями для синтеза белка Cas9, так же как и гидовые РНК, которые нацелены на определенные области ДНК.
Рассмотрим пример с мышами, болевшими МДД. Заболевание обусловлено мутациями в гене, который содержит инструкции для производства дистрофина. После проведения лечения системой CRISPR/Cas9, удалось восстановить функцию дистрофина в мышечных клетках мышей.
Похожие доклинические исследования проводятся для оценки потенциала системы CRISPR/Cas9 в лечении других типов мышечной дистрофии.
Для получения подробной информации — к ознакомлению доступны более 200 статей по CRISPR/cas-9 на сайте журнала Биомолекула https://biomolecula.ru/search/crispr
Генная терапия
Генная терапия, или перенос генов, относится к доставке генов в качестве терапевтических агентов. Поскольку гены содержат инструкции по синтезу белка, это прямо или косвенно будет считаться терапией для нервно-мышечных заболеваний. Так как, перенесенные гены потенциально могут продолжать продуцировать белок в течение некоторого времени, генная терапия может предложить более надежное решение, чем другие методы лечения. При этом, генная терапия сталкивается со многими техническими проблемами, а также с особым контролем, установленным регулирующими органами, такими как Управление по контролю за продуктами и лекарствами США (FDA), что не позволяет генной терапии быть клинически применимым методом в настоящее время.
Ключевыми проблемами являются доставка генов в целевую ткань, избегая при этом нежелательных тканей и нежелательного иммунного ответа на белки, полученные из новых генов, или на средства доставки новых генов.
Ученые при поддержке MDA создали уменьшенный рабочий ген дистрофина, который был протестирован у мальчиков с МДД. Хотя лечение казалось безопасным, некоторые из мальчиков испытывали нежелательный иммунный ответ на белок дистрофина, который ограничивал эффективность переноса гена. Этот иммунный ответ подвергается дальнейшему исследованию.
Причины дистрофии Дюшенна
Причиной появления заболевания является врожденный дефект половой X-хромосомы. Поражен тот ген, который отвечает за выработку в организме мышечного белка — дистрофина. Именно он является основой всех мышечных волокон на клеточном уровне. Вещество необходимо для корректного развития скелета, способности мышц сокращаться и расслабляться множество раз подряд.
При болезни Дюшенна белок отсутствует вовсе или является дефектным, то есть не выполняет свою функцию. Он заменяется жировой или соединительной тканью, из-за чего движение становится невозможным.
Заболевание имеет рецессивный тип наследования, сцепленный с X-хромосомой, и возникает при мутациях в обеих парных носителях. Патология поражает преимущественно мальчиков, поскольку в их генетическом наборе имеется одна X-хромосома. Если она дефектная, то болезнь настает.
Ингибирование миостатина
Блокирование белка миостатина с помощью белка фоллистатина, представляет собой стратегию, которая имеет потенциал для лечения МДД и, вероятно, многих других нервно-мышечных заболеваний. Мыши с похожим на МДД заболеванием, получавшие гены белка фоллистатина, показали общее увеличение массы тела и веса отдельных мышц. У обезьян, которым был выполнен перенос гена фоллистатина, были более сильные и крупные мышцы.
Стратегия, получившая значительную поддержку MDA, заключается в подавлении действия белка природного происхождения, ограничивающего рост мышц- миостатина. Исследователи надеются, что блокирование миостатина может позволить мышцам расти больше и сильнее.
Ингибиторы миостатина привлекают большое внимание сообщества исследователей нервно-мышечных заболеваний с тех пор, как несколько лет назад было обнаружено, что люди и животные с генетическим дефицитом миостатина имеют большие мышцы и хорошую силу без видимых побочных эффектов. В 2010 году исследование показало, что мыши, лишенные дистрофина и имеющие заболевание, подобное МДД, получили пользу от лечения «приманкой», которая «выманивала» миостатин из их мышц.
Затем биотехнологическая компания Acceleron Pharma разработала препарат на основе этой «приманки» и начала его тестирование при поддержке MDA у мальчиков с МДД. К сожалению, во время этого испытания возникли неожиданные проблемы безопасности, в результате чего Acceleron прекратил его в 2011 году.
Компания надеется решить эти проблемы безопасности и возобновить тестирование ACE-031 или модифицированной версии ACE-031.
На сайте Clinicaltrials.gov можно ознакомиться с результатами исследования препарата ACE — 031 для пациентов с МДД4.
Другие стратегии ингибирования миостатина, такие как инъекция генов для миостатин-блокирующего фоллистатина, также находятся на рассмотрении.
Стволовые клетки
Стволовые клетки — это клетки на самых ранних стадиях развития. Они могут превратиться в специальный тип клеток (например, мышечные или нервные клетки), или сохранять плюрипотентность — способность развиваться в любой из ряда различных типов клеток.
Трансплантация стволовых клеток предлагается в качестве лечения таких заболеваний, как мышечная дистрофия. На основе клеточной терапии предпринимались попытки стимулировать регенерацию мышц с надеждой на то, что стволовые клетки восстановят мышечную функцию и исправят патологию путем повторного синтеза мышц. Стволовые клетки рассматриваются как подходящий вариант для терапевтического применения из-за их способности к самовосстановлению и потенциала дифференцировки.
В 2006 году при поддержке исследователи при поддержке Ассоциации мышечных дистрофий (MDA) восстановили подвижность у двух собак и стабилизировали функцию у третьей, используя стволовые клетки, взятые из мышечных кровеносных сосудов.
В исследовании, опубликованном в 2007 году, европейская исследовательская группа успешно использовала комбинацию генетической коррекции и стволовых клеток для лечения мышей с МДД. Исследователи в этом исследовании извлекали генерирующие мышцу стволовые клетки из мышечной ткани и крови у людей с МДД, исправляли генетическую ошибку в генах дистрофина клеток, а затем вводили клетки мышам с дефицитом дистрофина. Клетки, полученные из мышц, вызывали лучшую регенерацию мышц, чем клетки, полученные из крови.
В 2010 году французские ученые при поддержке MDA сообщали, что они идентифицировали ранее неизвестный тип мышечных стволовых клеток, расположенных в промежутках между мышечными волокнами у мышей. Хотя все это еще на ранних стадиях исследований, есть надежда, что новые клетки, получившие название PICs, могут играть важную роль в регенерации и восстановлении мышц.
В этом же году по утверждениям исследователей считалось, что для формирования новой мышечной ткани сначала требуется контролируемый тип повреждения ДНК. Новое открытие расширило понимание ученых о том, как незрелые мышечные клетки становятся мышцами, и помогло им управлять этим процессом для лечения нескольких форм мышечной дистрофии.
Стволовые клетки продолжают оставаться основной областью исследований для специалистов и исследователей в сфере нервно-мышечных заболеваний. Некоторые продолжают изучать мышечные сателлитные клетки, тип стволовых клеток, присутствующих в мышечной ткани. Другие изучают различные типы клеток, которые способны пережить трансплантацию в мышцы и продуцировать желаемые белки. Кроме того изучаются сходства и различия в развитии скелетных мышц и жировой ткани.
В последние годы (с 2013 по 2020 гг) были получены обнадеживающие результаты лечения человека с использованием зрелых стволовых клеток. Так проф. Sharma при участии соавторов., в 2013 году изучали эффект внутримышечной аутотрансплантации мезенхимальных стволовых клеток костного мозга у 150 пациентов с мышечной дистрофией (имеются ввиду стволовые клетки. Через 12 месяцев наблюдения у пациентов наблюдалось увеличение мышечной силы и улучшение походки. Симптоматические и функциональные улучшения также наблюдались в 86,67% случаев: у шести пациентов снижен уровень жировой инфильтрации и выявлена регенерация мышц на снимках МРТ , а у девяти — выявлены положительные изменения электрической активности мышц на электронейромиографии (ЭНМГ).
Мезанхимальные стволовые клетки состоят из множества клеток, таких как гематопоэтические стволовые клетки, тканеспецифические клетки-предшественники, стромальные клетки и специализированные клетки крови на разных стадиях развития5. Эти клетки обладают способностью мобилизовать и оказывать свои репаративные эффекты в месте повреждения. Они способствуют неоваскуляризации и усиливают ангиогенез (образование сосудов), продуцируя сигнальные молекулы, такие как факторы роста эндотелия сосудов и факторы роста фибробластов (FGF2). Они также способствуют ремоделированию тканей, предотвращают апоптоз (отмирание клеток), уменьшают воспаление, высвобождают факторы роста и активируют сателлитные клетки. Это паракринные эффекты, которые могут помочь в достижении желаемого результата клеточной терапии6,7. Аутологичные мезенхимальные стволовые клетки костного мозга были использованы в этом случае, потому что они не имеют этических проблем, и его безопасность была установлена (не требуется донор, это клетки самого пациента).
Трансплантация стволовых клеток в нужное место мышечного тела, как правило, является основной практической трудностью. Внутривенное введение стволовых клеток, полученных из костного мозга, показало успешное возвращение стволовых клеток в поврежденные мышечные ткани на моделях животных; однако это также рискует разбавлением концентрации клеток. Мышечная дистрофия в основном воспринимается как заболевание мышц, с малым количеством свидетельств нервно-мышечных поражений. Дистрофин является частью структурного белка, обнаруженного в миелине, образующего клетки Шванна и нервы. Демиелинизация и дегенерация как изменения в нервах могут происходить с такими нарушениями в клетках. Поэтому были выбраны два различных способа трансплантации клеток: внутримышечный и интратекальный. Мезенхимальные клетки костного мозга вводили в двигательные точки целевых слабых мышц для восстановления иннервирующего нерва, а также мышц. Известно, что спинномозговая жидкость содержит факторы роста, которые помогают росту коркового эпителия и стимулируют васкуляризацию в нервной системе, поэтому он использовался в качестве разбавляющей среды.
Особый интерес вызывает клинический случай пациента с мышечной дистрофией Беккера, в отношении которого была проведена терапия с использованием мезанхемальных клеток костного мозга.
Результаты были обнадеживающими. После клеточной терапии пациент дважды наблюдался в стационаре через 3 и 9 месяцев. Спустя 3 месяца было отмечено улучшение двигательной функции верхних конечностей. Выполнение движений над головой требовало сравнительно меньших усилий. Наблюдалось двустороннее снижение жесткости и псевдогипертрофии икроножных мышц . Отмечались значительные улучшения в положении стоя и сидя, в способности держать равновесие. Баланс в положении стоя и при ходьбе улучшился. Частота падений при ходьбе заметно уменьшилась с 4-5 падений в месяц до 1 падения за 3 месяца. Характеристики дыхательной функции также улучшились: жизненная емкость легких (ЖЕЛ) (с 1250 мл до 1750 мл) и пиковая скорость выдоха (ПСВ) (с 290 мл до 360 мл).
Более подробно читайте запись «Эффективность клеточной терапии при прогрессирующей мышечной дистрофии Беккера»8.
Симптомы
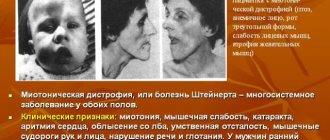
Старт заболевания приходится на возраст от 5 до 15 лет, но может быть и гораздо позже, после 40 лет. Первые признаки – повышенная утомляемость и мышечная слабость в области таза и нижних конечностей. У некоторых пациентов отмечаются периодические и возникающие спонтанно судороги икроножных мышц. Трудно подниматься по лестнице, вставать и садиться, при попытке встать со стула приходится искать точку опоры. Может иметь место так называемый симптом Говерса, когда человек как бы переступает руками по поверхности ног, помогая таким образом телу выпрямиться.
Боль и судороги в мышцах ног – характерный признак миопатии
Всем наследственным миопатиям свойственно симметричное развитие мышечной атрофии. Сначала поражаются бедренные мышцы и тазовой области, позже процесс переходит к мускулатуре плеч и верхней части рук. На начальных стадиях формируется псевдогипертрофия в икроножных мышцах, позже к ним присоединяются дельтовидная, трех- и четырехглавая мышца (квадрицепс бедра). В дальнейшем псевдогипертрофия трансформируется в мышечную гипотрофию.
Симптомы миопатии Беккера выражаются:
- нарушением сердечного ритма – аритмией;
- болью в мышцах рук и ног, а также других частей тела;
- болевые ощущения в ногах усиливаются при ходьбе;
- походка приобретает новые черты и становится похожей на утиную;
- в мышцах ног чувствуются постоянные подергивания;
- нормально передвигаться и обходить препятствия становится все труднее;
- даже незначительная физическая нагрузка сопровождается одышкой и усталостью, а также опуханием ног.
Полный комплекс симптомов редко присутствует у одного больного, однако утомляемость и слабость в ногах ощущают абсолютно все пациенты.
Особенность миопатии Беккера в том, что прогрессирует она достаточно медленно, но в любом случае приводит к обездвиженности и скованности суставов. Процесс продолжается примерно до 40 лет, по достижении этого возраста двигательная активность обычно прекращается. Следует отметить, что скелетных деформаций, например, искривления позвоночника, не происходит. Мозговая деятельность остается нормальной, сердце и сосуды поражаются в слабой степени. В некоторых случаях возможно наступление импотенции и снижение полового влечения, иногда наблюдается гинекомастия и атрофия яичек.
Усиление атрофина
Лабораторные данные показывают, что повышение уровня мышечного белка атрофина может, до некоторой степени, компенсировать дефицит дистрофина.
Утрофин очень похож на дистрофин, но, в отличие от дистрофина, обычно вырабатывается и полностью функционирует при МДБ. Следовательно, повышение уровня атрофина вряд ли вызовет нежелательный иммунный ответ, тогда как повышение уровня дистрофина может это сделать. Увеличение производства атрофина может помочь компенсировать дефицит дистрофина независимо от специфической мутации гена дистрофина.
Хотя по структуре и функции атрофин близок к дистрофину, между этими двумя белками есть как минимум одно ключевое отличие. Во время развития плода и, возможно, немного позднее, атрофин присутствует по всему мышечному волокну, взаимодействуя с кластерами белков, застрявшими в окружающей его мембране. По мере взросления животного или человека атрофин почти полностью заменяется дистрофином, за одним исключением. На нервно-мышечном соединении атрофин остается на протяжении всей жизни.
Несколько стратегий в настоящее время пытаются увеличить атрофин. Одним из них является выявление и подавление всего, что препятствует выработке атрофина — найти тормоз и, так сказать, отпустить его.
Другая стратегия заключается в том, чтобы ввести модифицированную версию самого белка атрофина в организм. Исследование, проведенное в 2009 году, показало, что модифицированный протеин атрофина дает значительные преимущества при введении мышам, у которых отсутствует белок дистрофина и которые имеют заболевание, напоминающее МДД.
В 2011 году ученые сообщили, что системное введение человеческой формы белка, называемого бигликан, мышам с болезнью, подобной МДД , повышает устойчивость мышечных мышц к повреждениям, связанным с их сокращением.
Биография
В 1894 году поступил в Школу военно-медицинской службы в Лионе. В 1897 году представил диссертацию на соискание докторской степени, посвящённую «антагонизму между плесенью и микробами».
Дюшен послал свою диссертацию в Институт Пастера, но не получил ответа. Он считал необходимым продолжение исследований, но этому помешала дальнейшая военная служба.
Один год провёл в интернатуре в Валь-де-Грас, затем был назначен майором медицины 2-го класса во 2-й гусарский полк в Санлис.
В 1901 году женился. Через два года жена умерла от туберкулёза.
В 1904 году заболел тяжёлой грудной болезнью (предположительно — туберкулёзом). Через три года был уволен с военной службы и направлен в санаторий в Амели-ле-Бен.
Умер 12 апреля 1912 года, в возрасте 37 лет. Похоронен рядом с женой на кладбище Гран-Жас в Каннах.